
Enhanced Sensitivity to Benzodiazepines in Active Cocaine-Abusing Subjects: A PET Study
Abstract
OBJECTIVE: Because cocaine enhances dopamine brain activity and dopamine signals are transferred through γ-aminobutyric acid pathways, the authors hypothesized GABA-ergic disruption in cocaine-abusing subjects. This study tests this hypothesis. METHOD: GABA brain function was assessed indirectly by measuring the brain metabolic responses to lorazepam, a drug that facilitates GABA neurotransmission. Thirteen current cocaine-abusing subjects and 14 comparison subjects were scanned twice with positron emission tomography and [18F]fluorodeoxyglucose; the first scan was obtained after placebo administration and the second after lorazepam administration (30 µg/kg). RESULTS: Despite significantly higher plasma lorazepam concentrations in comparison subjects than in cocaine-abusing subjects, lorazepam-induced decrements in whole brain metabolism were significantly greater in cocaine-abusing (mean=21%, SD=13%) than in comparison (mean=13%, SD=7%) subjects. These differences were largest in striatum, thalamus, and parietal cortex. Lorazepam-induced sleepiness in cocaine-abusing subjects was intense and was significantly greater than in comparison subjects, and it was correlated with lorazepam-induced changes in thalamic metabolism. Whereas regional metabolic measures during placebo administration were significantly higher in cocaine-abusing subjects than in comparison subjects, the measures during lorazepam administration were equivalent for both groups. CONCLUSIONS: The enhanced sensitivity to lorazepam in cocaine-abusing subjects suggests disruption of GABA pathways that may reflect, in part, cocaine withdrawal. The intense sleepiness induced by lorazepam in some of the abusers, despite their significantly lower plasma concentrations, should alert clinicians of the potential toxicity from accentuated responses to sedative hypnotics in active cocaine-abusing subjects. (Am J Psychiatry 1998; 155:200–206)
Although the ability of cocaine to increase dopamine in nucleus accumbens has been shown to be crucial for cocaine reinforcement (1, 2), the role of dopamine in the loss of control and the compulsive self-administration of cocaine in the cocaine addict is much less clear. It has been postulated that the dopamine system's involvement in addiction is by means of its disruption of the brain regions that it modulates (3). The expression of dopamine-mediated behaviors requires the activation of γ-aminobutyric acid (GABA) pathways (4), which are therefore a particularly susceptible target for cocaine's effects. In fact, using positron emission tomography (PET), we have documented significant reductions in striatal dopamine D2 receptors in cocaine abusers that persist after detoxification (5, 6). Because dopamine D2 receptors predominantly reflect postsynaptic elements on GABA cells (7), these findings suggest an involvement of GABA in the dopamine abnormalities in cocaine abusers. In the present study we evaluated the GABA system indirectly by assessing the brain regional responsivity to GABA stimulation and compared the responses in cocaine-abusing subjects to those in comparison subjects.
Responsivity to GABA stimulation was quantified by measuring the regional brain metabolic response to lorazepam by using [18F]fluorodeoxyglucose (FDG) and PET (8). Benzodiazepines have been used to assess the response of the brain to GABA stimulation since they facilitate GABA neurotransmission (9, 10). Benzodiazepines produce a pattern of changes in regional brain metabolism that parallels the regional concentration of benzodiazepine receptors in brain (11) and that is reversed by the benzodiazepine antagonist flumazenil (12). Metabolic responses to benzodiazepines are highly reproducible in a given subject and are consistent across normal individuals (13).
The regional brain metabolic responses to lorazepam were measured in 13 active cocaine-abusing subjects and compared to those in 14 healthy subjects. Because dopamine is inhibitory to striatal GABA cells (4), we hypothesized that chronic cocaine abusers would have decreased GABA function with a concomitant receptor sensitization that would manifest itself as an enhanced response to lorazepam.
METHOD
Subjects
Thirteen current cocaine-abusing subjects (mean age=38 years, SD=6) were selected from a pool of cocaine-abusing subjects who responded to an advertisement. The inclusion criteria were 1) meeting DSM-IV criteria for active cocaine dependence, 2) continuous use of cocaine for at least the prior 6 months and claimed cocaine use of at least “three grams” a week (estimated cost of $300/week), and 3) use of cocaine as smoked free-base and/or intravenously. Exclusion criteria were current or past psychiatric disease (other than cocaine dependence); neurological signs and/or history of neurological disease; history of head trauma with loss of consciousness; cardiovascular or endocrinological disease; current medical illness; dependence on any substance other than cocaine, nicotine, or caffeine; and more than moderate (12 ounces/week) use of ethanol. Evaluation of subjects was performed consistently by the same clinician (G.-J.W.), and diagnoses were separately confirmed (N.D.V.).
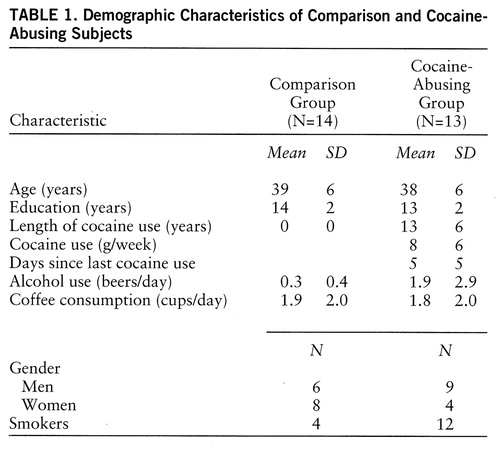
The comparison group comprised 14 healthy volunteers (mean age=39 years, SD=6) who were screened for a lack of current or past psychiatric or neurological disease, as well as for a lack of drug or alcohol abuse (excluding nicotine and caffeine). Exclusion criteria were otherwise the same as those used for the cocaine-abusing group. None of the subjects in the comparison or the cocaine-abusing group was taking medications at the time of the study. Table 1 provides the demographic characteristics of the subjects.
As part of the evaluation procedure, subjects had physical, psychiatric, and neurologic examinations. Routine laboratory tests, as well as a random urine test, were performed to exclude the use of psychoactive drugs other than cocaine in the abusing group. Subjects were instructed to refrain from drinking alcohol the week before the PET scan. Cigarettes, food, and beverages were discontinued at least 4 hours before the study. Written informed consent was obtained from each subject after the procedure was fully explained. Subjects were monetarily remunerated ($100 per day of participation) when the studies were completed.
Scanning
PET measurements were done by using a CTI 931 tomograph (6.5×5.9×5.9 mm full-width half maximum). Details on subject positioning, catheterizations, transmission and emission scans, radiotracer synthesis, and calculation of metabolic “rates” have been published elsewhere (14). Briefly, emission scans were taken 35 minutes after injection of 4–6 mCi of FDG for a total of 20 minutes. Arterialized blood was obtained throughout the procedure to measure plasma concentrations of F-18, glucose, PO2, pCO2, and lorazepam. Lorazepam concentration in plasma was measured 20, 50, 70, and 95 minutes after lorazepam administration with high-performance liquid chromatography (National Psychopharmacology Laboratory).
Each subject underwent two PET FDG scans obtained within 1 week of each other. The first FDG scan was obtained after intravenous administration of placebo (3 cc of saline solution) given 40–50 minutes before FDG; and the second after intravenous administration of lorazepam (30 µg/kg) given 40–50 minutes before FDG. The subjects were blind to the drugs received. To ensure that the subjects would not fall asleep, they were monitored throughout the procedure and were asked to keep their eyes open. Subjects were scanned with their ears unplugged in a dimly lit room with noise kept to a minimum. Lorazepam-induced sleepiness and its effects on cognitive tasks were periodically assessed as previously described (12).
Data Analysis
Regions of interest were drawn directly on the transaxial PET images through use of a template that selected 72 regions of interest from 10 of the 15 axial planes. Weighted averages of the regions of interest from different slices corresponding to the same anatomical areas were computed to obtain metabolic values in nine “composite” brain regions. The template indicating size and location of the region of interest sampled and the regions of interest that were included to obtain the composite regions have been published elsewhere (15).
Differences in regional metabolism between the cocaine-abusing group and the comparison group were tested with analysis of variance (ANOVA). Differences in regional brain metabolic responses to lorazepam between the groups were tested by using the differences between placebo and lorazepam with one-factor (diagnosis) repeated (regions) measures ANOVA. Post hoc t tests were then performed to assess the brain regions when these differences were significant. In consideration of the “multiple comparison problem” incurred by analyzing values for nine brain regions in the post hoc t tests, we set the level of significance to p≤0.01. We chose this criterion of significance as being intermediate between the p≤0.05 value, considered significant for an individual variable, and the p<0.0056 value required by the Bonferroni adjustment since Bonferroni criterion assumes that variables are independent (16), but regional metabolic values are highly dependent on one another (17).
Pearson product moment correlation analysis was used to assess the relationship between changes in regional metabolism and lorazepam-induced sleepiness and plasma lorazepam concentration at different times after its administration. The level of significance for the correlations (nine regions and four plasma measurements) was set at p≤0.005. We chose this criterion of significance as being intermediate between the p≤0.05 value, considered significant for an individual variable, and the p<0.0014 value required by the Bonferroni adjustment.
Differences in self-ratings of sleepiness after placebo and after lorazepam administration and differences in plasma lorazepam concentrations between the groups were tested with repeated measure ANOVA.
RESULTS
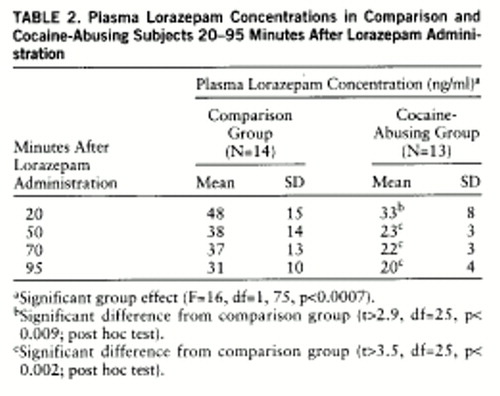
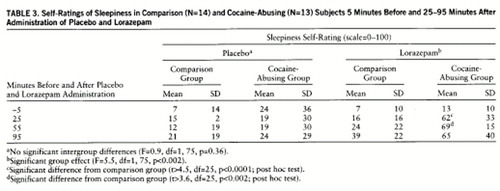
Plasma lorazepam concentrations were significantly higher in the comparison than in the cocaine-abusing group (group effect: F=16, df=1,75, p<0.0007) (table 2). Despite higher plasma levels of lorazepam in the comparison group, reports of lorazepam-induced sleepiness were significantly higher in the cocaine-abusing group (interaction effect: F=5.5, df=1,75, p≤0.002) (table 3). We do not report cognitive effects of lorazepam, since we were unable to assess them properly in the cocaine-abusing group because of the extreme and prolonged levels of sleepiness induced by lorazepam in some of these subjects. The ANOVA for the placebo condition showed no differences in self-reports of sleepiness between groups (F=0.9, df=1,25, p=0.36) (table 3).
Lorazepam decreased global and regional brain glucose metabolism, and these decrements were significantly larger in the cocaine abuser than in the comparison group (figure 1). Whereas in the comparison group whole brain metabolism decreased 13% (SD=7%), in the cocaine-abusing group it decreased 21% (SD=3%) (F=6.2, df=1,25, p<0.02). The results from the ANOVA obtained on the difference scores for the regional metabolic measures showed a significant overall difference between the abuser and the comparison groups (F=5.8, df=1,25, p<0.03), as well as a significant group-by-region interaction effect (F=2.8, df=1,200, p<0.006). The presence of a significant group-by-region interaction effect indicates that the lorazepam-induced differences in metabolic activity between the comparison and the abuser groups differed across brain regions. Post hoc t tests showed that these differences were significant in striatum (t=2.7, df=25, p<0.01), thalamus (t=2.8, df=25, p<0.01), and parietal cortex (t=2.8, df=25, p<0.01) (figure 2). Lorazepam-induced increases in self-ratings of sleepiness obtained 25 and 55 minutes after lorazepam administration were significantly correlated with the decreases in thalamic metabolism (r=0.67, df=25, p<0.0001, and r=0.65, df=25, p<0.0002) (figure 3). There were no significant correlations between plasma lorazepam concentration and lorazepam-induced metabolic changes.
At baseline, whole brain metabolism was significantly higher in the cocaine-abusing group than in the comparison group (ANOVA, group effect: F=7.5, df=1,25, p<0.01), whereas after lorazepam administration, regional metabolic measures were equivalent for the abuser and the comparison groups (figure 4). Post hoc t tests showed that differences in baseline metabolic measures were significantly higher in cocaine-abusing subjects in the occipital cortex (t=2.8, df=25, p<0.01), temporal cortex (t=3.2, df=25, p<0.005), thalamus (t=3.9, df=25, p<0.0006), and striatum (t=4, df=25, p<0.0004).
DISCUSSION
This study documents an increased sensitivity to lorazepam in active cocaine-abusing subjects as assessed by the enhanced responses to its sedative effects and to its reduction of brain glucose metabolism. Although the accentuated brain metabolic responses were global, there were some brain regions where these effects were more pronounced than others, as shown by the significant group-by-region interaction effect. More specifically, the differences in responses between the abuser and the comparison groups were largest in thalamus, striatum, occipital cortex, and parietal cortex. Because metabolism reflects activity in terminal regions (18), the metabolic changes induced by lorazepam in a given area predominantly reflect the responses in the projecting GABA cells. Thus, one could speculate that the increased metabolic response to lorazepam in thalamus in the cocaine abusers could reflect increased GABA sensitivity in pallidum and in substantia nigra reticulata (4). However, it could also reflect changes in other GABA-ergic populations (i.e., GABA interneurons). We interpret these findings as reflecting adaptation changes in the GABA system secondary to intermittent cocaine intoxication.
The regional specificity for the enhanced responses in the cocaine abusers is likely to reflect the sequential as well as the parallel organization of GABA output systems (19). Thus, adaptive processes in response to dopamine stimulation are likely to depend on the brain regions and the balance between them (20), as well as the subtype of GABA and/or dopamine receptors that prevail in a given region (19). In addition, regional metabolic responses to benzodiazepines probably reflect not only direct interactions with benzodiazepine receptors but also secondary effects. The extent to which the enhanced response to lorazepam reflects differences in receptors or secondary effects cannot be determined by this study. Future imaging studies using benzodiazepine receptor ligands coupled to metabolic studies with a benzodiazepine challenge are required to clarify this issue.
Studies in laboratory animals have shown that chronic cocaine affects the GABA system, and this includes changes in GABA receptors (function, concentration, composition, and transduction elements) (21–23) and in enzymes that regulate GABA synthesis (24). In general, most studies document enhancement of GABA-ergic responses with chronic cocaine administration, including an enhanced sensitivity to the sedative effects of benzodiazepines (25). However, results are not always consistent. For example, while most binding studies have reported increases in striatal concentration of GABA receptors (26, 27), others have reported decreases (28). Similarly, while some studies of chronically treated animals have reported an enhanced sensitivity to iontophoretically applied GABA (29, 30), others have reported decreases (31). These inconsistencies could reflect the different adaptation responses in GABA cells depending on whether they receive direct or indirect dopamine projections. Inconsistencies could also reflect differences in the regimens of cocaine administration used as well as the time after withdrawal at which measurements were made (25, 27). Although most of these studies have linked the changes in the GABA system to the process of sensitization (32), their role in cocaine addiction per se has not been evaluated.
Studies of cocaine abusers have also shown evidence of changes in the GABA system. However, as with the laboratory animal literature, there are few studies, and the pattern of abnormalities has not been consistent. For example, while some studies have reported increases in peripheral benzodiazepine receptors in platelets of cocaine abusers (33), others have shown decreases (34). The phenomenology associated with cocaine intoxication and withdrawal is also suggestive of an involvement of GABA. For example, convulsions associated with cocaine intoxication respond to benzodiazepine agonists but not to the anticonvulsant phenytoin (35). In addition, cocaine intoxication is associated with anxiety, agitation, and, not infrequently, panic attacks, which are symptoms that respond to benzodiazepines (36, 37). Furthermore, the frequent combined use of alcohol and cocaine by cocaine-abusing subjects (38) could reflect in part a means to compensate for cocaine-induced decreases in GABA activity, since alcohol potentiates GABA-induced chloride flux at the GABA receptor (39, 40). In this respect, it was interesting to note that the increased metabolic activity at baseline observed in the cocaine abusers, which we had previously reported to be related to days since last cocaine use (41), returned to values similar to those in the comparison group after lorazepam administration. Thus, one could postulate that this metabolic hyperactivity observed during early detoxification may reflect decreased GABA-ergic activity shortly after cocaine discontinuation. Although in laboratory animals discontinuation of chronic low doses of alcohol can produce global increases in glucose metabolism (42), alcohol consumption in the abusers is unlikely to account for the baseline metabolic differences between groups, since the amount of alcohol they reported they used was comparable to that reported by the comparison subjects (table 1). However, because histories of alcohol and drug use are not always reliable and we had no laboratory corroboration, we cannot exclude the contribution of variables other than cocaine in the hypermetabolic baseline measures observed in the abusers.
In cocaine-abusing subjects brain glucose metabolism decreases after protracted withdrawal (43), and thus further studies are required to determine if the enhanced responsivity to lorazepam in the abusers also decreases as a function of withdrawal or whether it represents a more lasting adaptation (or predisposing factor), as seen for the changes in dopamine D2 receptors that persist after detoxification (6).
The finding of an enhanced sensitivity to lorazepam in cocaine-abusing subjects differs markedly from the findings reported in alcoholic subjects, in whom a blunted response to lorazepam-induced changes in regional brain metabolism has been reported (14). Because subjects at risk for alcoholism do not show the blunted brain metabolic response to lorazepam (8), the response in the alcoholics was interpreted as reflecting the effects of alcohol administration and/or alcohol withdrawal. In this study we excluded subjects who abused alcohol, and thus future studies comparing response to lorazepam between cocaine-abusing subjects who abuse alcohol and those who do not are required to determine the generalizability of our findings in cocaine-abusing subjects.
The lower plasma lorazepam concentration in the cocaine-abusing group than in the comparison group is most likely due to enhanced metabolism of lorazepam and may reflect hepatic enzymatic induction in cocaine-abusing subjects, since lorazepam is metabolized by glucuronide conjugation in the liver (44). The mechanisms accounting for the enzyme induction are unclear and could reflect the use of other substances (i.e., alcohol, cigarettes, other sedative/hypnotic drugs) in the cocaine-abusing subjects. In addition, because cocaine can be metabolized by glucuroconjugation (45), the possibility of a direct effect of cocaine on glucuronyl transferase should be further investigated. Because the enhanced sensitivity to lorazepam in cocaine-abusing subjects was observed even in the presence of much lower plasma lorazepam concentrations, the potential toxicity of sedative hypnotic drugs for which there is no enhanced metabolism and/or for which the safety is not as good as for benzodiazepines should be further evaluated. This increased sensitivity, for example, could contribute to the enhanced toxicity and mortality observed with the combined use of alcohol and cocaine (46). In addition, the extreme sedative effects observed for some of the cocaine-abusing subjects after lorazepam administration should alert the clinician of potential untoward reactions with the use of these drugs by active cocaine-abusing subjects in situations where alertness is required.
In summary, these data document an accentuated sensitivity to benzodiazepines in cocaine-abusing subjects, which appears as an enhanced response to the sedative effects of lorazepam and an enhanced decrease in regional brain metabolism relative to that in nonabusing subjects. These results support the notion of disruption of GABA activity in the brain of cocaine-abusing subjects. Further studies are required to determine the extent to which these changes are due to cocaine withdrawal and/or prolonged sleep deprivation and whether they will decrease and/or disappear with protracted detoxification.
 |
 |
 |
Received Jan. 9, 1997; revision received April 22, 1997; accepted May 2, 1997. From the Medical and Chemistry Departments, Brookhaven National Laboratory; and the Department of Psychiatry, State University of New York at Stony Brook, Stony Brook, N.Y. Address reprint requests to Dr. Volkow, Medical Department, Brookhaven National Laboratory, Bldg. 490, Upton, NY 11973; [email protected] (e-mail). Supported in part by contract DE-ACO2-76 CH-00016 from the Office of Health and Environmental Research, U.S. Department of Energy, and grant DA-06891 from the National Institute on Drug Abuse. The authors thank David Alexoff, Robert Carciello, Richard Ferrieri, Payton King, Alex Levy, Robert MacGregor, Noelwah Netusil, Carol Redvanly, David Schlyer, Colleen Shea, Donald Warner, and Cristopher Wong for advice and assistance.

FIGURE 1. Images of Brain Glucose Metabolism for a Comparison Subject and a Cocaine-Abusing Subject After Placebo and After Lorazepam Administrationa
aLorazepam decreased metabolism, and the changes were larger in the cocaine-abusing subject than in the comparison subject.

FIGURE 2. Regional Changes in Metabolism Induced by Lorazepam in Comparison and in Cocaine-Abusing Subjectsa
aLorazepam-induced decrements in regional brain glucose metabolism were significantly larger in the cocaine-abusing group than in the comparison group (ANOVA, group effect: F=5.8, df=1,25, p<0.03).
bt>2.8, df=25, p<0.01; post hoc t tests.

FIGURE 3. Correlation Between Lorazepam-Induced Changes in Regional Metabolic Activity in Thalamus and in Self-Reports of Sleepiness 25 Minutes After Its Administration for Comparison (N=14) and Cocaine-Abusing (N=13) Subjectsa
ar=0.67, df=25, p<0.0001. Measures correspond to the differences between scores obtained before and those obtained after lorazepam administration.

FIGURE 4. Regional Brain Metabolic Measures in Comparison Subjects and in Cocaine-Abusing Subjects After Placebo and After Lorazepam Administrationa
aBaseline metabolic measures were significantly higher in cocaine-abusing subjects than in comparison subjects during placebo (ANOVA, group effect: F=7.5, df=1,25, p<0.01), whereas regional metabolic measures were equivalent between the groups during lorazepam administration.
bt=3.2, df=25, p<0.005; post hoc t tests.
ct=2.8, df=25, p<0.01; post hoc t tests.
dt>3.9, df=25, p<0.0006; post hoc t tests.
1. Koob GF, Bloom FE: Cellular and molecular mechanisms of drug dependence. Science 1988; 242:715–723Crossref, Medline, Google Scholar
2. Di Chiara G, Imperato A: Drugs abused by humans preferentially increase synaptic dopamine concentrations in the mesolimbic system of freely moving rats. Proc Natl Acad Sci USA 1988; 85:5274–5278Google Scholar
3. Le Moal M, Simon H: Mesocorticolimbic dopaminergic network: functional and regulatory notes. Physiol Rev 1991; 71:155–234Crossref, Medline, Google Scholar
4. Scheel-Krüger J: Dopamine-GABA interactions: evidence that GABA transmits, modulates and mediates dopaminergic functions in the basal ganglia and the limbic system. Acta Neurol Scand 1986; 73:9–49Crossref, Google Scholar
5. Volkow ND, Fowler JS, Wolf AP, Shlyer D, Shiue C-Y, Albert R, Dewey SL, Logan J, Bendriem B, Christman D, Hitzemann R, Henn F: Effects of chronic cocaine abuse on postsynaptic dopamine receptors. Am J Psychiatry 1990; 147:719–724Link, Google Scholar
6. Volkow ND, Fowler JS, Wang GJ, Hitzemann R, Logan J, Schlyer D, Dewey SL, Wolf AP: Decreased dopamine D2 receptor availability is associated with reduced frontal metabolism in cocaine abusers. Synapse 1993; 14:169–177Crossref, Medline, Google Scholar
7. Volkow ND, Fowler JS, Gatley JS, Logan J, Wang G-J, Ding Y-S, Dewey SL: Evaluation of the human brain dopamine system with PET. J Nucl Med 1996; 37:1242–1256Google Scholar
8. Volkow ND, Wang GJ, Begleiter H, Hitzemann R, Pappas N, Burr G, Pascani K, Wong C, Fowler JS, Wolf AP: Regional brain metabolic response to lorazepam in subjects at risk for alcoholism. Alcohol Clin Exp Res 1995; 19:510–516Crossref, Medline, Google Scholar
9. Olsen RW: GABA benzodiazepine barbiturate receptor interactions. J Neurochem 1981; 37:1–3Crossref, Medline, Google Scholar
10. Morrow AL, Paul SM: Benzodiazepine enhancement of gamma aminobutyric acid-mediated chloride ion flux in rat brain synaptoneurosomes. J Neurochem 1988; 50:302–306Crossref, Medline, Google Scholar
11. Buchsbaum M, Wu J, Haier R, Hazlett E, Ball R, Katz M, Sokolski K, Lagunas-Solar M, Langer D: Positron emission tomography, assessment of effects of benzodiazepines on regional glucose metabolism. Life Sci 1987; 40:2393–2400Google Scholar
12. Volkow ND, Wang GJ, Hitzemann R, Fowler JS, Pappas N, Loremier P, Burr G, Pascani K, Wong C, Wolf AP: Depression of thalamic metabolism by lorazepam is associated with sleepiness. Neuropsychopharmacology 1995; 12:123–132Crossref, Medline, Google Scholar
13. Wang GJ, Volkow ND, Overall JE, Hitzemann RJ, Pappas N, Pascani K, Fowler JS: Reproducibility of regional brain metabolic responses to lorazepam in normal human subjects. J Nucl Med 1996; 37:1609–1613Google Scholar
14. Volkow ND, Wang G-J, Hitzemann R, Fowler JS, Wolf AP, Pappas N, Biegon A, Dewey SL: Decreased cerebral response to inhibitory neurotransmission in alcoholics. Am J Psychiatry 1993; 150:417–422Link, Google Scholar
15. Wang G-J, Volkow ND, Roque C, Cestaro V, Hitzemann R, Cantos E, Levy AV, Wolf AP: Functional significance of ventricular enlargement and cortical atrophy in normals and alcoholics as assessed by PET, MRI and neuropsychological testing. Radiology 1992; 186:59–65Crossref, Google Scholar
16. Hays WL: Statistics for the Social Sciences. New York, Holt, Reinhart & Winston, 1973Google Scholar
17. Volkow ND, Brodie JD, Wolf AP, Gomez-Mont F, Cancro R, Van Gelder P, Russell JAG, Overall J: Brain organization of schizophrenics. J Cereb Blood Flow Metab 1986; 6:441–446Crossref, Medline, Google Scholar
18. Schwartz WJ, Smith CB, Davidsen L, Savaki H, Sokoloff L, Mata M, Fink DJ, Gainer H: Metabolic mapping of functional activity in the hypothalamo-neurohypophysial system of the rat. Science 1979; 205:723–725Crossref, Medline, Google Scholar
19. Yoshida M: The GABAergic systems and the role of the basal ganglia in motor control. Adv Biochem Pharmacol 1981; 30:37–52Medline, Google Scholar
20. Walter JR, Pucak ML: The modulation of midbrain dopaminergic systems by GABA, in The Modulation Of Midbrain Dopaminergic Neurotransmission by Other Neurotransmitters. Edited by Ashby CR Jr. Boca Raton, Fla, CRC Press, 1996, pp 157–187Google Scholar
21. Goeders NE: Cocaine differentially affects benzodiazepine receptors in discrete regions of the rat brain: persistence and potential mechanisms mediating these effects. J Pharmacol Exp Ther 1991; 259:574–581Medline, Google Scholar
22. Carney DA, Abel MS: Repeated cocaine treatment produces selective alterations in mRNA for subunits of the GABAA receptors. Abstracts of the Society for Neuroscience 1995; 21:714Google Scholar
23. Nestler EJ, Terwilliger RZ, Walker JR, Sevarino KA, Duman RS: Chronic cocaine treatment decreases levels of the G protein subunits Gia and Goa in discrete regions of rat brain. J Neurochem 1990; 55:1079–1082Google Scholar
24. Sorg BA, Guminski BJM, Hooks MS, Kalivas PW: Cocaine alters glutamic acid decarboxylase differentially in the nucleus accumbens core and shell. Brain Res Mol Brain Res 1995; 29:381–386Crossref, Medline, Google Scholar
25. Lipton JW, Ellison GD: Continuous cocaine induces persistent changes in behavioral responsivity to both scopolamine and diazepam. Neuropsychopharmacology 1992; 7:143–148Medline, Google Scholar
26. Zeigler S, Lipton J, Toga A, Ellison G: Continuous cocaine administration produces persisting changes in brain neurochemistry and behavior. Brain Res 1991; 552:27–35Crossref, Medline, Google Scholar
27. Lipton JW, Olsen RW, Ellison GD: Length of continuous cocaine exposure determines the persistence of muscarinic and benzodiazepine receptor alterations. Brain Res 1995; 676:378–385Crossref, Medline, Google Scholar
28. Pecins-Thompson M, Peris J: Behavioral and neurochemical changes caused by repeated ethanol and cocaine administration. Psychopharmacology (Berl) 1993; 110:443–450Crossref, Medline, Google Scholar
29. Henry DJ, White FJ: The persistence of behavioral sensitization to cocaine parallels enhanced inhibition of nucleus accumbens neurons. J Neurosci 1995; 15:6287–6298Google Scholar
30. Waterhouse BD, Stowe ZN, Jimenez-Rivera CA, Sessler FM, Woodward DJ: Cocaine actions in a central noradrenergic circuit: enhancement of cerebellar Purkinje neuron responses to iontophoretically applied GABA. Brain Res 1991; 546:297–309Crossref, Medline, Google Scholar
31. Peris J: Repeated cocaine injections decrease the function of striatal γ-aminobutyric acidA receptors. J Pharmacol Exp Ther 1996; 276:1002–1008Google Scholar
32. Steketee JD, Kalivas PW: Sensitization to psychostimulants and stress after injection of pertussis toxin into the A10 dopamine region. J Pharmacol Exp Ther 1991; 259:916–924Medline, Google Scholar
33. Chesley SF, Scharzki AD, De Urrutia J, Greenblatt DJ, Shader RI, Miller LG: Cocaine augments peripheral benzodiazepine binding in humans. J Clin Psychiatry 1990; 51:404–406Medline, Google Scholar
34. Javaid JI, Notorangelo MP, Pandey SC, Reddy PL, Pandey GN, Davis JM: Peripheral benzodiazepine receptors are decreased during cocaine withdrawal in humans. Biol Psychiatry 1994; 36:44–50Crossref, Medline, Google Scholar
35. Tarr JE, Macklin M: Cocaine. Pediatr Clin North Am 1987; 34:319–331Crossref, Medline, Google Scholar
36. Aronson TA, Craig TJ: Cocaine precipitation of panic disorder. Am J Psychiatry 1986; 143:643–645Link, Google Scholar
37. Cowley DS: Alcohol abuse, substance abuse, and panic disorders. Am J Med 1992; 92:41–48Crossref, Medline, Google Scholar
38. Grant BF, Harford TC: Concurrent and simultaneous use of alcohol with cocaine: results of national survey. Drug Alcohol Depend 1990; 25:97–104Crossref, Medline, Google Scholar
39. Ticku MK, Mhatre M, Metha AK: Modulation of GABAergic transmission by ethanol. Adv Biochem Psychopharmacol 1992; 47:255–268Medline, Google Scholar
40. Kulonen E: Ethanol and GABA. Med Biol 1983; 61:247–167Google Scholar
41. Volkow ND, Fowler JS, Wolf AP, Hitzemann R, Dewey S, Bendriem B, Alpert R, Hoff A: Changes in brain glucose metabolism in cocaine dependence and withdrawal. Am J Psychiatry 1991; 148:621–626Link, Google Scholar
42. Williams-Hemby L, Grant KA, Gatto GJ, Porrino LJ: Metabolic mapping of the effects of chronic voluntary ethanol consumption in rats. Pharmacol Biochem Behav 1996; 54:415–423Crossref, Medline, Google Scholar
43. Volkow ND, Hitzemann R, Wang G-J, Fowler JS, Wolf AP, Dewey SL: Long-term frontal brain metabolic changes in cocaine abusers. Synapse 1992; 11:184–190Crossref, Medline, Google Scholar
44. Rall TW: Hypnotics and sedatives: ethanol, in Goodman and Gilman's The Pharmacological Basis of Therapeutics, 8th ed. Edited by Gilman AG, Rall TW, Nies AS, Taylor P. New York, Pergamon Press, 1990, pp 345–382Google Scholar
45. Boelsterli UA, Goldlin C: Biomechanisms of cocaine-induced hepatocyte injury mediated by the formation of reactive metabolites. Arch Toxicol 1991; 65:351–360Crossref, Medline, Google Scholar
46. Kreek MJ, Stimmel B: Alcoholism and Polydrug Use. New York, Haworth Press, 1984Google Scholar

