
Priming the Brain for Psychosis: Maternal Inflammation During Fetal Development and the Risk of Later Psychiatric Disorder
Thirty years ago, in a cafe in Helsinki, Sarnoff Mednick and Matti Huttunen contemplated the conundrum of the season-of-birth effect in schizophrenia. Why were individuals with schizophrenia more likely to be born in winter or early spring than in summer? Inspired by earlier ideas by Fuller Torrey and others, they speculated that prenatal viral infection may be involved, and they devised a test of this hypothesis using the known dates of the 1957 influenza pandemic in Uusimaa County, Finland. Heading to the psychiatric hospital archives where case notes were stored by date of birth, they noticed that the proportion of patients with schizophrenia diagnoses appeared higher among those born in the spring of 1958, who would have been in mid-gestation during the epidemic, when compared with those born later in 1958 or in preceding or subsequent years. Their paper, building on these original observations, was published in 1988 (1) and was highly influential in promoting a prenatal viral infection theory of schizophrenia. Although the methodology of the original paper has proved controversial (2), the findings were, to a greater or lesser extent, replicated by O’Callaghan et al. (3) and many others, extending to other infectious agents and culminating in a comprehensive review by Brown and Derkits (4), which made the startling assertion that 30% of schizophrenia cases could be prevented if certain prenatal infections were entirely eliminated from the pregnant population.
Any prenatal infection theory of schizophrenia etiology must take account of the fact that most infections do not reach the fetus itself, and it has long been considered that the mother’s inflammatory response may be a key factor linking early-life insults to later disorder. In this issue of the Journal, the article by Canetta et al. (5), from Alan Brown’s research group, provides the most robust evidence to date that maternal inflammation is a risk factor for later schizophrenia, using unique data drawn from the Finnish Maternity Cohort, an impressive total birth cohort of over one million pregnancies in Finland since 1983. Stored first-trimester and early second-trimester serum samples were available from 777 individuals who had received diagnoses of schizophrenia from the Finnish Hospital Discharge Register up to 1998 and from matched controls. Canetta et al. report that very high maternal levels of C-reactive protein, an acute phase protein that acts as a marker of both acute and chronic inflammatory conditions, were associated with a nearly 60% increased risk of later schizophrenia diagnosis. This risk also extended across the spectrum of inflammation, with a 28% increase in risk for schizophrenia with every unit increase in C-reactive protein.
What Are the Implications of These Findings?
Firstly, while the authors only assessed C-reactive protein, proinflammatory markers, such as interleukin 8 and tumor necrosis factor alpha, have been shown to have similar associations in other birth cohorts (6). This suggests that the risk is associated with a generally elevated inflammatory state. Secondly, the inflammation story does not appear to be specific to schizophrenia because elevated markers of inflammation are also found in association with depression, with posttraumatic stress disorder, and in many physical diseases (7) Indeed, Brown and his research group have also reported elevated levels of maternal C-reactive protein in association with autism (8).
What is Causing the Inflammatory Process?
Maternal infection is an obvious causal candidate, but inflammation may represent a common pathway mediating a diverse range of pregnancy-related and early-childhood exposures previously associated with schizophrenia (9, 10). Evidence of increased inflammation has been found in neonates exposed to obstetric complications, such as preterm labor, preeclampsia, and hypoxia, and in pregnant mothers exposed to psychosocial stress (11). As Mednick et al. (1) pointed out in their 1988 study, “It seems likely that a variety of illnesses or physical or psychological stresses at this critical point in gestation can have related effects on brain development.” However, the window of risk may also extend beyond infancy. Early-life adversity, such as childhood trauma or bullying, can lead to elevated levels of inflammatory markers (12, 13) and are risk factors for psychosis and other mental disorders (14). The inflammatory system and the hypothalamic-pituitary-adrenal (HPA) axis, which mediates the stress response, are inextricably linked: cytokines can elicit a stress response through activation of the fetal HPA axis, and stressors can lead to HPA axis dysregulation and loss of normal glucocorticoid-associated anti-inflammatory tone (15). Cotter and Pariante (16) previously proposed that many of the neuropathological features observed in schizophrenia are in keeping with nonspecific glucocorticoid-related brain changes. Autoimmunity may also underlie some of the elevation in inflammatory tone seen in people with schizophrenia, and a bidirectional association between schizophrenia and autoimmune disorders has been reported (17). The recent upsurge of interest in anti-N-methyl-d-aspartic acid receptor encephalitis as a differential diagnosis for schizophrenia demonstrates the importance of autoimmunity in psychosis (18). It is also possible that only a subset of subjects with schizophrenia show elevated inflammatory markers and that there may be a pathophysiologically distinct subtype of schizophrenia related to immune regulation.
How Does It All Fit Together?
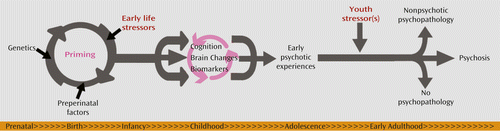
Canetta et al. report a small but robust dose-response relationship between prenatal inflammatory markers and later schizophrenia. It is possible, though unlikely, that the association is one of simple cause and effect, where a single prenatal insult is sufficient in itself to lead to elevated proinflammatory cytokines, altered brain development, and subsequent psychiatric disorder. Urs Meyer and his research group (19) have proposed a “two-hit” model, based on animal work, whereby an early infection, or other inflammatory process, “primes” the developing brain, but the onset of disorder requires a second stressor during the pubertal period. Translated to human terms, we could speculate that exposure to an elevated inflammatory state in prenatal life or early childhood (for example, as a result of prenatal infection, maternal stress, early-life adversity, or obstetric exposures), and possibly in association with a genetic vulnerability toward dysregulation of the immune system (20), may “prime” the brain and lead to vulnerability. This vulnerability state may be expressed, for example, clinically as early psychotic symptoms (21), cognitively as subtle speed of processing deficits (22), or neurologically as disrupted brain connectivity through altered synaptic pruning (23). Further stressors during youth development, such as adolescent bullying, assault, stressful life transitions, relationship breakups, or substance use, may serve to convert the vulnerability into disorder (14). These ideas are presented graphically in Figure 1.
What Else Do We Need to Know?
Further focus is needed on the longitudinal dimension of neuroinflammation to address the possibility of a chronic inflammatory process underlying schizophrenia. Inflammatory markers have been shown, in separate studies, to be elevated both preceding and following the onset of the disorder (6). However, it is not known whether these changes reflect one-off alterations in different subjects at different times or relapsing changes within the same subjects. Ideally, one would need prospectively collected inflammatory markers from the prenatal period through childhood and adolescence until illness onset to fully establish the direction of the association between inflammation and schizophrenia and to identify mediators and moderators along the causal pathway. We need to know more about the combinations and timing of risk factors that propel an individual exposed to maternal inflammation along a pathway that may lead to psychosis rather than depression or autism. The study of interaction or synergy between risk factors in psychiatric disorders needs further attention. We previously reported that a synergistic association between genetic risk and maternal infection could account for up to 46% of schizophrenia illness among exposed cases (24).
What Are the Implications for Treatment and Prevention?
The protection of pregnant women from infection and stress might at first glance seem to be beyond the province of most psychiatrists. Indeed, infection during pregnancy is an increasing concern of obstetricians, since recent evidence indicates that pregnant women are particularly vulnerable to certain infections (25). Nevertheless, stress management, and treatment of depression and anxiety, does encompass the responsibilities of psychiatrists who work with pregnant women. Comprehensive psychiatric and psychological treatment for expectant mothers, as well as physical monitoring, would now seem indicated not only for the health of the mother but also to thereby decrease the longer-term risk for mental illness in her child.
1 : Adult schizophrenia following prenatal exposure to an influenza epidemic. Arch Gen Psychiatry 1988; 45:189–192Crossref, Medline, Google Scholar
2 : Evidence against maternal influenza as a risk factor for schizophrenia. Br J Psychiatry 1994; 164:674–676Crossref, Medline, Google Scholar
3 : Schizophrenia after prenatal exposure to 1957 A2 influenza epidemic. Lancet 1991; 337:1248–1250Crossref, Medline, Google Scholar
4 : Prenatal infection and schizophrenia: a review of epidemiologic and translational studies. Am J Psychiatry 2010; 167:261–280Link, Google Scholar
5 : Elevated maternal C-reactive protein and increased risk of schizophrenia in a national birth cohort. Am J Psychiatry 2014; 171:960–968Link, Google Scholar
6 : Inflammation theories in psychotic disorders: a critical review. Infect Disord Drug Targets 2013; 13:59–70Crossref, Medline, Google Scholar
7 : Inflammatory biomarkers profiles of mental disorders and their relation to clinical, social and lifestyle factors. Soc Psychiatry Psych Epidemiol 2014; 49:841–849Crossref, Medline, Google Scholar
8 : Elevated maternal C-reactive protein and autism in a national birth cohort. Mol Psychiatry 2014; 19:259–264Crossref, Medline, Google Scholar
9 : Obstetric complications and schizophrenia: historical and meta-analytic review. Am J Psychiatry 2002; 159:1080–1092Link, Google Scholar
10 : Sudden death of father or sibling in early childhood increases risk for psychotic disorder. Schizophr Res 2013; 143:363–366Crossref, Medline, Google Scholar
11 : Prenatal inflammation and neurodevelopment in schizophrenia: a review of human studies. Prog Neuropsychopharmacol Biol Psychiatry 2013; 42:92–100Crossref, Medline, Google Scholar
12 : Biological embedding of stress through inflammation processes in childhood. Mol Psychiatry 2011; 16:244–246Crossref, Medline, Google Scholar
13 : Childhood bullying involvement predicts low-grade systemic inflammation into adulthood. Proc Natl Acad Sci USA 2014; 111:7570–7575Crossref, Medline, Google Scholar
14 : Childhood trauma and psychosis in a prospective cohort study: cause, effect, and directionality. Am J Psychiatry 2013; 170:734–741Link, Google Scholar
15 : Cytokines and the neurodevelopmental basis of mental illness. Front Neurosci (Epub ahead of print, Oct 17, 2013)Google Scholar
16 : Stress and the progression of the developmental hypothesis of schizophrenia. Br J Psychiatry 2002; 181:363–365Crossref, Medline, Google Scholar
17 : A nationwide study on the risk of autoimmune diseases in individuals with a personal or a family history of schizophrenia and related psychosis. Am J Psychiatry 2014; 171:218–226Link, Google Scholar
18 : Anti-NMDA receptor encephalitis: an important differential diagnosis in psychosis. Br J Psychiatry 2011; 199:508–509Crossref, Medline, Google Scholar
19 : Stress in puberty unmasks latent neuropathological consequences of prenatal immune activation in mice. Science 2013; 339:1095–1099Crossref, Medline, Google Scholar
20 : Genome-wide association studies: findings at the major histocompatibility complex locus in psychosis. Biol Psychiatry 2014; 75:276–283Crossref, Medline, Google Scholar
21 : Psychotic-like experiences in the general population: characterizing a high-risk group for psychosis. Psychol Med 2011; 41:1–6Crossref, Medline, Google Scholar
22 : Neurocognition in the extended psychosis phenotype: performance of a community sample of adolescents with psychotic symptoms on the MATRICS neurocognitive battery. Schizophr Bull 2013; 39:1018–1026Crossref, Medline, Google Scholar
23 : The complement system: an unexpected role in synaptic pruning during development and disease. Annu Rev Neurosci 2012; 35:369–389Crossref, Medline, Google Scholar
24 : Evidence for an interaction between familial liability and prenatal exposure to infection in the causation of schizophrenia. Am J Psychiatry 2009; 166:1025–1030Link, Google Scholar
25 : Pregnancy and infection. N Engl J Med 2014; 370:2211–2218Crossref, Medline, Google Scholar

