
Genome-Wide Linkage and Follow-Up Association Study of Postpartum Mood Symptoms
Abstract
Objective: Family studies have suggested that postpartum mood symptoms might have a partly genetic etiology. The authors used a genome-wide linkage analysis to search for chromosomal regions that harbor genetic variants conferring susceptibility for such symptoms. The authors then fine-mapped their best linkage regions, assessing single nucleotide polymorphisms (SNPs) for genetic association with postpartum symptoms. Method: Subjects were ascertained from two studies: the NIMH Genetics Initiative Bipolar Disorder project and the Genetics of Recurrent Early-Onset Depression. Subjects included women with a history of pregnancy, any mood disorder, and information about postpartum symptoms. In the linkage study, 1,210 women met criteria (23% with postpartum symptoms), and 417 microsatellite markers were analyzed in multipoint allele sharing analyses. For the association study, 759 women met criteria (25% with postpartum symptoms), and 16,916 SNPs in the regions of the best linkage peaks were assessed for association with postpartum symptoms. Results: The maximum linkage peak for postpartum symptoms occurred on chromosome 1q21.3-q32.1, with a chromosome-wide significant likelihood ratio Z score (Z LR ) of 2.93 (permutation p=0.02). This was a significant increase over the baseline Z LR of 0.32 observed at this locus among all women with a mood disorder (permutation p=0.004). Suggestive linkage was also found on 9p24.3-p22.3 (Z LR =2.91). In the fine-mapping study, the strongest implicated gene was HMCN1 (nominal p=0.00017), containing four estrogen receptor binding sites, although this was not region-wide significant. Conclusions: This is the first study to examine the genetic etiology of postpartum mood symptoms using genome-wide data. The results suggest that genetic variations on chromosomes 1q21.3-q32.1 and 9p24.3-p22.3 may increase susceptibility to postpartum mood symptoms.
Mood symptoms and syndromes are common during and after pregnancy and are potentially harmful to both mother and infant. While genetic factors clearly influence mood disorders in general, evidence for a genetic role specific to postpartum symptoms is less well-established, as this area has been little explored. Postpartum depression occurs in up to 10%–20% of mothers in the year following delivery (1 , 2) . The risk for postpartum depression is increased in women with a history of major depression (3 , 4) and in women with a history of postpartum depression following previous pregnancies (5 , 6) . Our group found that approximately 20% of women with major depression report depressive symptoms in the month following delivery (7) . In women with bipolar disorder, postpartum mood episodes including both depression and mania have been reported at rates as high as 25%–50% (8) ; the specific risk of postpartum psychosis, a syndrome resembling mania with psychotic features, is 20%–30% (9 – 11) , although it remains unclear if prophylactic treatment lowers this rate. We have shown that approximately 20% of women with bipolar disorder report significant mood symptoms within a month of childbirth and that approximately 50% experience significant symptoms either during pregnancy or postpartum (7) .
A genetic basis for postpartum mood syndromes is suggested by several studies. Family studies of postpartum psychosis have supported a genetic susceptibility to a postpartum trigger in bipolar disorder, as well as an overlap in genetic factors predisposing to postpartum psychosis and bipolar disorder (12 , 13) . Dean et al. (14) found a higher risk of postpartum mood illness in relatives of probands with postpartum psychosis. Forty et al. (15) showed that the trait of postpartum depression exhibited familiality in pedigrees with recurrent major depression. We have reported familial aggregation of postpartum depressive symptoms in families with recurrent early onset major depression and bipolar disorder (16 , 17) . There has been one genome-wide linkage study related to postpartum mood symptoms, narrowly focused on postpartum psychosis (18) . To date there has been no linkage study of postpartum mood symptoms nor one focused on depressive symptoms in this setting.
We therefore undertook a genome-wide linkage scan of postpartum mood symptoms in pedigrees with major depression or bipolar disorder. We made use of large genotyped family sets available for both disorders through two collaboratives. We further had the benefit of data from genome-wide association studies conducted by the collaborative groups, with which we fine-mapped our linkage findings.
Method
Subjects
The data for this study was collected by two efforts, the NIMH Genetics Initiative Bipolar Disorder project (NIMH-BP) and the Genetics of Recurrent Early-Onset Major Depression study (GenRED). Eligibility, ascertainment, and assessment procedures for waves 3 and 4 of the NIMH-BP sample have been described elsewhere (19 , 20) . Subjects from waves 1 and 2 were excluded because complete information about peripartum symptoms was unavailable. Families had a proband with bipolar I disorder and at least one other sibling with bipolar I disorder or schizoaffective disorder, bipolar type. After complete description of the study, written informed consent was obtained. Family members were assessed with the Diagnostic Interview for Genetic Studies (DIGS) (21) . This was combined with medical record and family informant data to assign diagnoses based on DSM-III-R or DSM-IV criteria. For the fine-mapping study, the sample included probands from the families plus unrelated bipolar I disorder cases ascertained as part of NIMH-BP wave 5.
GenRED I was a family study for which methods have been previously described (22 , 23) . Families were ascertained if the proband and at least one sibling had early onset major depression. Early onset was defined as onset before age 31 in probands and before age 41 in relatives. Written informed consent was obtained after complete description of the study. The DIGS was employed and DSM-IV diagnoses were assigned. GenRED II (24) ascertained unrelated major depression cases who had an affected parent or sibling through assessment with the Family Interview for Genetic Studies. For the fine-mapping study, major depression cases from the GenRED II sample were included along with probands from the families.
We restricted our study to women who had a history of pregnancy and any best-estimate mood disorder diagnosis. Additionally, we used responses from two sections of the DIGS interview to determine whether women had postpartum mood symptoms. One section queried: “Have you ever had any severe emotional problems during a pregnancy or within a month of childbirth?” Affirmative answers were classified as during pregnancy, both during and after pregnancy, or after pregnancy. The second asked whether the most severe depression occurred during or after a pregnancy. Women were considered to have postpartum mood symptoms if they endorsed either of these two items for the time period after pregnancy only.
For the linkage analysis, the majority of subjects reported being Caucasian (92%), with a minority black (3%) or other/unknown (5%). In the association analysis, 98.7% were self-reported as Caucasian. Ninety NIMH-BP subjects and 218 GenRED subjects were included in both the linkage and association analyses.
Microsatellite Genotyping
The NIMH-BP families used in the linkage analyses were genotyped genome-wide in two waves (waves 3 and 4) at the Center for Inherited Disease Research of Johns Hopkins (http://www.cidr.jhmi.edu), as described previously (19 , 20) . We used PREST (http://www.stat.uchicago.edu/∼mcpeek/software/prest/) on the genotype data to verify reported familial relationships, UNKNOWN (25) and/or PEDCHECK (26) to identify Mendelian inheritance errors, and CRIMAP (http://compgen.rutgers.edu/multimap/crimap/) to identify unlikely double recombinations. Data identified as potentially erroneous were deleted if unable to be resolved. Similarly, the GenRED families were genotyped in two waves at the Center for Inherited Disease Research, as described previously (27) . Error checking of the genotype data were performed using RELCHECK and PEDCHECK (26 , 28) to identify and remove Mendelian inconsistencies and SIMPED (http://www.hgsc.bcm.tmc.edu/genemapping) to exclude genotypes with an estimated probability of error exceeding 70%.
We combined the cleaned genotype data from the two NIMH-BP waves and the two GenRED waves into a single dataset and made a common genetic map with all available markers. The deCode genetic map was used as a framework (29) , with interpolation from Marshfield locations (http://research.marshfieldclinic.org/genetics). Markers not available from either of these sources were placed on the framework according to their relative physical position from the July 2003 assembly of the human genome sequence (http://genome.ucsc.edu). We then interpolated their genetic locations based on their physical position relative to the nearest flanking markers with known genetic locations. For markers genotyped in multiple waves, we placed all instances of the marker next to each other on the map with an intermarker distance of 0.01. There were 1,210 women from 495 families who met our inclusion criteria and were informative for linkage. Genotypes were available at 1,575 markers (417 unique markers), spaced at ∼9 cM across the genome.
SNP Genotyping
The NIMH-BP SNP genotyping was performed genome-wide on bipolar disorder cases and unrelated controls as part of the Genetic Association Information Network (GAIN) Bipolar Initiative (30) . In a separate effort, genome-wide SNP genotyping was performed on major depression cases from the GenRED sample (24) . Genotyping in both samples was performed using the Affymetrix Genome-wide Human SNP Array 6.0. Each study removed subjects with low call rates, plate effects, Mendelian errors, or low heterozygosity. We performed additional quality control on the SNP data from each sample separately, removing SNPs with minor allele frequency <0.01, missing data rate>0.05, Hardy-Weinberg Equilibrium p<0.000001, and plate effects. We then performed another round of quality control on the SNPs, removing SNPs with minor allele frequency or missing data rates differing between the two samples (p<0.001). While genotyping and initial quality control was performed genome-wide, the focus of our current study was restricted to a 49.75-Mb region on chromosome 1 and a 14.34-Mb region on chromosome 9. Thus, we kept only those SNPs in our regions of interest that were successfully genotyped and passed quality control in both samples. This resulted in a final dataset of 759 subjects meeting our inclusion criteria genotyped at 11,557 SNPs on chromosome 1q21.3-q32.1 and 5,359 SNPs on chromosome 9p24.3-p22.3.
Linkage Analysis
For a baseline analysis, we included as affected all women with a history of pregnancy and any mood disorder diagnosis. A separate analysis was performed on the subgroup of women who had postpartum mood symptoms in addition to the baseline criteria. We analyzed the genome-wide microsatellite data using a multipoint allele sharing model in Allegro 2.0 (31) . The analysis considered all possible informative affected relative pairs, weighted by a function that is approximately halfway between weighting by each family versus by each pair, and generated 10 steps between genotyped markers. Allele frequencies were calculated using the entire sample from both NIMH-BP and GenRED (6,573 subjects). We empirically tested for genome-wide significance of our linkage peaks under the postpartum mood symptom model by simulating 1,000 genome-wide datasets, shuffling the genotype, and counting the number of times that a likelihood ratio Z score (Z LR ) more significant than the observed Z LR was found across the genome. We then tested whether the linkage peak was unique to the postpartum mood symptom model versus the baseline model by simulating 1,000 genome-wide datasets, permuting the phenotype label of postpartum mood symptoms among women meeting baseline criteria, and counting the number of times that a Z LR more significant than our observed Z LR was found across the genome. Genome-wide significance was defined as a simulated result more extreme than the observed result occurring in fewer than 5% of the simulated genome scans.
Association Analysis
We conducted a SNP association analysis in the 2-Z LR regions surrounding our linkage peaks on chromosome 1 and 9. We employed a case-only strategy among women who met our baseline criteria, comparing women with postpartum mood symptoms to those without postpartum mood symptoms. We used both meta-analytic and mega-analytic approaches to analyze NIMH-BP and GenRED samples together. First, we conducted meta-analyses of the data, combining the test statistics for each SNP across samples. In each sample separately, we performed a case-only analysis using the LOGISTIC routine in PLINK (32) and an additive genotype model counting the number of rare alleles. The genomic inflation factor in NIMH-BP sample was 1.03 and in the GenRED sample it was 1.12. Since the GenRED data contained significant population substructure, we chose to adjust for substructure in each sample using principal components. Using EIGENSTRAT (33) , we identified 10 principal components for the GenRED sample and two principal components for the GAIN sample. We incorporated covariates for each principal component for the specific sample into the logistic regression model. After calculating the association for each sample separately, we utilized two different meta-analytic approaches. First, we used a weighted Z-score analysis (34) . For each SNP, we converted the p value for association into a Z score. We calculated a Z meta score as the sum of the Z scores from each study, weighted by the effective sample size, and found the associated p value. The second approach was a random effects meta-analysis. We performed this analysis using the META routine in STATA 9.0, calculating pooled effect estimates and confidence intervals (35) . We used a Bonferroni correction to adjust for the multiple SNPs tested in the regions. We tested for between-study heterogeneity in effects using Cochran’s Q.
We then combined the data from NIMH-BP and GenRED samples at the genotype level in a mega-analysis. We performed a case-only analysis using the LOGISTIC routine in PLINK, testing whether the association for each SNP differed between women with and without postpartum mood symptoms and using an additive genotype model counting the number of rare alleles. We did not correct for population stratification in the combined dataset to prevent against overconservative estimates of the effect size. We empirically tested the region-wide significance of our association findings using the MPERM routine in PLINK with label-swapping and 1,000 permutations. Any SNPs that passed our initial quality control yet still had relatively high missing data rate (>3%) or differential missingness by phenotype or by genotype (p<0.075) were carefully checked due to the potential for a false positive result. We tested for evidence of interaction using the epistasis procedure in PLINK, assessing the interaction of each SNP in our top gene of interest with every other SNP in the second strongest gene.
Results
Linkage
The final linkage dataset included genotypes from 1,210 informative women, 757 from GenRED and 453 from NIMH-BP. Complete information about the timing of mood symptoms in relation to childbirth was not available for 84 subjects. Of the remaining 1,126 subjects, 258 (22.9%) had postpartum mood symptoms. The mood disorder diagnoses present in this sample included recurrent major depression (62.6%), bipolar I disorder (26.9%), single-episode major depression (3.9%), bipolar II disorder (3.4%), schizoaffective disorder-bipolar type (1.3%), and other mood disorders (1.9%). There were no differences between the NIMH-BP and GenRED samples in mean age at onset of mood disorder (20.5 and 20.0 years, respectively; t=0.91, p=0.3633), mean age at interview (44.3 and 45.0 years; t= –1.01, p=0.3129) or rate of postpartum mood symptoms (21.4% and 23.6%; χ 2 =0.70, p=0.4019).
Table 1 shows the numbers and types of informative affected relative pairs for all women in the baseline group and for the subset with postpartum mood symptoms. There were 961 informative affected relative pairs for the baseline linkage analysis and 63 for the analysis among the subset with postpartum mood symptoms. Figure 1 illustrates the results for our top four chromosomal regions, based on the maximum Z LR score in the linkage analysis for the subset with postpartum mood symptoms. Complete genome-wide results are provided in Supplementary Figure 1 that accompanies the online version of this article. A maximum Z LR of 2.93 was observed in the postpartum mood symptom group for marker D1S1660 at 189.3 cM on chromosome 1. Permutation testing showed this result was chromosome-wide significant (p=0.02), although it did not reach genome-wide significance (p=0.35). At this location, the Z LR in the baseline group was only 0.32, so the increase from baseline was 2.61 in the postpartum mood symptom group (empirical p=0.004). The 2-Z LR support region around the linkage peak included the chromosomal region 1q21.3-q32.1, spanning 60 cM and containing seven markers genotyped across all samples. The mean information content value in the postpartum mood symptoms analysis was 0.715 genome-wide and 0.783 in the 2-Z LR region. Other linkage peaks (maximum Z LR >2.0) in the postpartum mood symptoms analysis were seen on chromosomes 2q37.1-q37.3, 9p24.3-p22.3, and 13q21.33-q33.1 ( Figure 1 ). However, of these regions, only a 29-cM region on 9p24.3-p22.3 showed a significant difference between the postpartum mood symptom group and the baseline group (change in Z LR =2.77, empirical p=0.001).

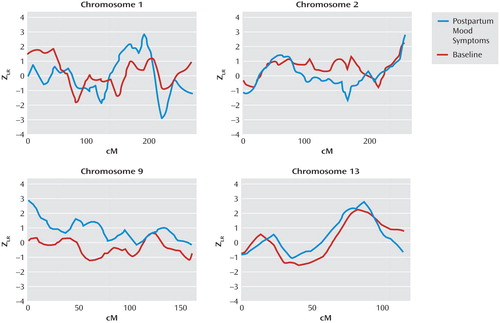
a Z LR are the Z likelihood ratio score statistics from linkage analyses of the baseline phenotype of all women with a pregnancy and a mood disorder and for the subphenotype of women with postpartum mood symptoms. No region yielded genome-wide significant evidence of linkage.
Association
Encouraged by the linkage specific to postpartum mood symptoms on chromosomes 1q21.3-q32.1 and 9p24.3-p22.3, we sought to follow-up these findings in a densely genotyped SNP association study. Of the 759 women who met our baseline criteria and had genotype data available for this analysis, 457 were from GenRED and 302 from NIMH-BP. The distribution of mood disorder diagnoses was as follows: recurrent major depression, 58.8%; bipolar I disorder, 38.7%; single-episode major depression, 1.4%; and schizoaffective disorder-bipolar type, 1.1%. The NIMH-BP subjects were slightly older at interview than the GenRED subjects (45.2 versus 43.6 years, respectively; t=1.98, p=0.0483). There was no difference in the proportion of women with postpartum mood symptoms between the NIMH-BP (25.2%) and GenRED (24.3%) samples (χ 2 =0.08, p=0.7838). Overall, 187 women (24.6%) met our criteria for postpartum mood symptoms.
Our top case-only meta-analysis and mega-analysis association results are summarized in Table 2 , which lists all results with p<0.001 using any method. All results from our two linkage regions are presented in Supplementary Figure 2 . Our best finding was on chromosome 1 for SNP rs16852397, with a mega-analytic odds ratio of 1.98 (p=3.58×10 –4 ). This SNP was also nominally associated with postpartum mood symptoms in the meta-analytic methods controlling for population stratification. rs16852397 is intergenic but is located in a spliced EST. Our best association signal on chromosome 9 was for the intergenic SNP rs7025259 (meta-analytic p=9.56×10 –4 ). Two genes that were implicated among our top findings are HMCN1 (Hemicentin 1) and METTL13 (Methyltransferase like 13) on chromosome 1 ( Figure 2 ). None of these findings were significant after accounting for the multiple SNPs tested across the regions. For our top results, there was no evidence of between-sample heterogeneity (Q-statistic p>0.05). We tested whether SNPs in our two top genes of interest showed evidence of interaction in relation to postpartum mood symptoms. The strongest interaction between two SNPs across these genes was nominally significant (p=0.0027). This does not hold up to multiple testing given that 924 comparisons were made between 66 SNPs in HMCN1 and 14 SNPs in METTL13 .
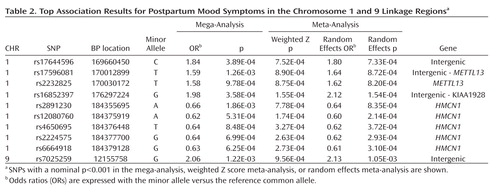
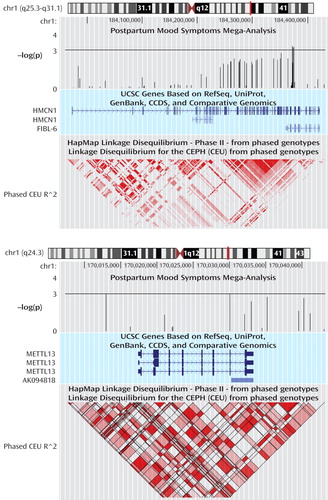
a Shown are the cytogenic region, -log(p values) for the association mega-analysis, UCSC genes, and the linkage disequilibrium structure in the region. Locations are based on NCBI build 36 (UCSC release March 2006).
Discussion
To our knowledge, this is the first study to examine the genetic etiology of postpartum mood symptoms using a systematic, genome-wide approach. We observed genome-wide suggestive linkage signals on 1q21.3-q32.1 and 9p24.3-p22.3 that may be specific to postpartum mood symptoms. We followed up these 2-Z LR regions surrounding the linkage peaks on chromosomes 1 and 9 using a SNP association study. Our best association signal on chromosome 1 was for the intergenic SNP rs16852397 and on chromosome 9 was for the intergenic SNP rs7025259. We also found modest evidence of association for SNPs on chromosome 1 in the genes HMCN1 and METTL13 with postpartum mood symptoms.
While ours is the first genome-wide linkage study of postpartum mood symptoms, one previous study examined linkage in postpartum psychosis (18) . Using the Wellcome Trust UK-Irish Bipolar Sib-pair sample and analyzing only families in which at least one woman had an episode of postpartum psychosis, Jones et al. reported genome-wide significant linkage on chromosome 16p13 and suggestive linkage on 8q24. Neither of these regions reached nominal significance in our study. It is worth emphasizing that in our study less than 30% of the subjects were diagnosed with bipolar I disorder, and that our definition of postpartum mood symptoms was much broader than that used by Jones et al. Our best linkage peak on chromosome 1 was previously reported to show modest evidence of linkage in Ashkenazi Jewish bipolar disorder families (36) . This finding is consistent with our observation of a modest linkage signal in this region in our baseline group, and an enhanced signal in the group with postpartum mood symptoms. No evidence of linkage with mood disorders has been previously reported in our region on chromosome 9p.
Motivated by the hypothesis that peripartum mood syndromes may have a unique genetic etiology, several studies have examined candidate genes for association with peripartum psychosis. Polymorphisms in the serotonin receptor gene HTR2C and the serotonin transporter SLC6A4 have been found to be associated with peripartum psychosis (37) . However, no associations have been reported with polymorphisms in the genes ESR1, HTR2A, NR3C1, and TNFA(37 – 41) . HMCN1 and METTL13 have not been previously examined for association with peripartum mood syndromes.
HMCN1 is 456 kb long and codes for an extracellular matrix protein with several functions, including transmembrane receptor activity and calcium ion binding. The gene contains four experimentally determined estrogen receptor binding sites (42) , which might be relevant for a postpartum phenotype. HMCN1 is particularly highly expressed in the hippocampus (43) , a brain region likely to be involved in depression (44) , and shown to be altered in rats by a postpartum drop in estrogen levels (45) . Twenty-seven SNPs in HMCN1 were at least nominally significant (p<0.05), suggesting our results are not due to genotyping error. Our genotyped SNPs in HMCN1 passing quality control captured 83% of the common variation in the gene (MAF≥0.01, r 2 ≥0.8). The gene METTL13 is putatively involved with methyltransferase activity and is 16 kb in length. Interestingly, DNA methyltransferases have been shown to play a role in estrogen receptor-induced gene transcription (46) . Six of the available SNPs passing quality control in METTL13 were at least nominally significant. The genotyped SNPs captured 88% of the common variation in this gene.
Our results should be viewed in light of several limitations. First, these samples were originally ascertained for other purposes. While the clinical data were collected using a rigorous and well validated instrument, the interview did not contain all possible information about peripartum mood symptoms. Second, we combined samples from different sources for this study, potentially introducing heterogeneity. We combined these samples to create one of the largest datasets available to examine the genetic etiology of postpartum mood symptoms. We felt this was appropriate as the samples used similar ascertainment, assessment, and genotyping methods. Third, prospective data might provide greater clarity for assessing the timing of the onset of symptoms relative to parturition. Fourth, we corrected the meta-analysis for population stratification but not the mega-analysis, as it can lead to overconservative estimates of effect sizes. This may have contributed to the slight differences observed between the results from these two approaches. Finally, even though we combined samples to create a larger dataset, we still had limited power to detect loci of modest effect.
In conclusion, we performed the first genome-wide linkage analysis of postpartum mood symptoms using a large sample with detailed clinical information. We followed up our best linkage peaks in an association study with densely genotyped SNPs. Our results suggest there may be genetic variation contributing to susceptibility to postpartum mood symptoms in the 1q21.3-q32.1 and 9p24.3-p22.3 regions. Specifically, the genes HMCN1 and METTL13 may contain polymorphisms that confer susceptibility to postpartum mood symptoms. As both the linkage and association results presented here are novel, future studies replicating these findings are warranted.
1. Campbell SB, Cohn JF: Prevalence and correlates of postpartum depression in first-time mothers. J Abnorm Psychol 1991; 100:594–599Google Scholar
2. Robinson GE, Stewart DE: Postpartum psychiatric disorders. CMAJ 1986; 134:31–37Google Scholar
3. Frank E, Kupfer DJ, Jacob M, Blumenthal SJ, Jarrett DB: Pregnancy-related affective episodes among women with recurrent depression. Am J Psychiatry 1987; 144:288–293Google Scholar
4. Kumar R, Robson KM: A prospective study of emotional disorders in childbearing women. Br J Psychiatry 1984; 144:35–47Google Scholar
5. Cooper PJ, Campbell EA, Day A, Kennerley H, Bond A: Non-psychotic psychiatric disorder after childbirth: a prospective study of prevalence, incidence, course and nature. Br J Psychiatry 1988; 152:799–806Google Scholar
6. Cox JL, Murray D, Chapman G: A controlled study of the onset, duration and prevalence of postnatal depression. Br J Psychiatry 1993; 163:27–31Google Scholar
7. Payne JL, Roy PS, Murphy-Eberenz K, Weismann MM, Swartz KL, McInnis MG, Nwulia E, Mondimore FM, Mackinnon DF, Miller EB, Nurnberger JI, Levinson DF, Depaulo JR Jr, Potash JB: Reproductive cycle-associated mood symptoms in women with major depression and bipolar disorder. J Affect Disord 2007; 99:221–229Google Scholar
8. Hunt N, Silverstone T: Does puerperal illness distinguish a subgroup of bipolar patients? J Affect Disord 1995; 34:101–107Google Scholar
9. Bratfos O, Haug JO: Puerperal mental disorders in manic-depressive females. Acta Psychiatr Scand 1966; 42:285–294Google Scholar
10. Kendell RE, Chalmers JC, Platz C: Epidemiology of puerperal psychoses. Br J Psychiatry 1987; 150:662–673Google Scholar
11. Reich T, Winokur G: Postpartum psychoses in patients with manic depressive disease. J Nerv Ment Dis 1970; 151:60–68Google Scholar
12. Jones I, Middle F, McCandless F, Coyle N, Robertson E, Brockington I, Lendon C, Craddock N: Molecular genetic studies of bipolar disorder and puerperal psychosis at two polymorphisms in the estrogen receptor alpha gene (ESR 1). Am J Med Genet 2000; 96:850–853Google Scholar
13. Jones I, Craddock N: Familiality of the puerperal trigger in bipolar disorder: results of a family study. Am J Psychiatry 2001; 158:913–917Google Scholar
14. Dean C, Williams RJ, Brockington IF: Is puerperal psychosis the same as bipolar manic-depressive disorder? A family study. Psychol Med 1989; 19:637–647Google Scholar
15. Forty L, Jones L, Macgregor S, Caesar S, Cooper C, Hough A, Dean L, Dave S, Farmer A, McGuffin P, Brewster S, Craddock N, Jones I: Familiality of postpartum depression in unipolar disorder: results of a family study. Am J Psychiatry 2006; 163:1549–1553Google Scholar
16. Murphy-Eberenz K, Zandi PP, March D, Crowe RR, Scheftner WA, Alexander M, McInnis MG, Coryell W, Adams P, Depaulo JR Jr, Miller EB, Marta DH, Potash JB, Payne J, Levinson DF: Is perinatal depression familial? J Affect Disord 2006; 90:49–55Google Scholar
17. Payne JL, Mackinnon DF, Mondimore FM, McInnis MG, Schweizer B, Zamoiski RB, McMahon FJ, Nurnberger JI Jr, Rice JP, Scheftner W, Coryell W, Berrettini WH, Kelsoe JR, Byerley W, Gershon ES, Depaulo JR Jr, Potash JB: Familial aggregation of postpartum mood symptoms in bipolar disorder pedigrees. Bipolar Disord 2008; 10:38–44Google Scholar
18. Jones I, Hamshere M, Nangle JM, Bennett P, Green E, Heron J, Segurado R, Lambert D, Holmans P, Corvin A, Owen M, Jones L, Gill M, Craddock N: Bipolar affective puerperal psychosis: genome-wide significant evidence for linkage to chromosome 16. Am J Psychiatry 2007; 164:1099–1104Google Scholar
19. Dick DM, Foroud T, Flury L, Bowman ES, Miller MJ, Rau NL, Moe PR, Samavedy N, El-Mallakh R, Manji H, Glitz DA, Meyer ET, Smiley C, Hahn R, Widmark C, McKinney R, Sutton L, Ballas C, Grice D, Berrettini W, Byerley W, Coryell W, DePaulo R, Mackinnon DF, Gershon ES, Kelsoe JR, McMahon FJ, McInnis M, Murphy DL, Reich T, Scheftner W, Nurnberger JI Jr: Genomewide linkage analyses of bipolar disorder: a new sample of 250 pedigrees from the National Institute of Mental Health Genetics Initiative. Am J Hum Genet 2003; 73:107–114Google Scholar
20. Kassem L, Lopez V, Hedeker D, Steele J, Zandi P, McMahon FJ: Familiality of polarity at illness onset in bipolar affective disorder. Am J Psychiatry 2006; 163:1754–1759Google Scholar
21. Nurnberger JI Jr, Blehar MC, Kaufmann CA, York-Cooler C, Simpson SG, Harkavy-Friedman J, Severe JB, Malaspina D, Reich T: Diagnostic interview for genetic studies: rationale, unique features, and training. NIMH Genetics Initiative. Arch Gen Psychiatry 1994; 51:849–859Google Scholar
22. Holmans P, Zubenko GS, Crowe RR, Depaulo JR Jr, Scheftner WA, Weissman MM, Zubenko WN, Boutelle S, Murphy-Eberenz K, MacKinnon D, McInnis MG, Marta DH, Adams P, Knowles JA, Gladis M, Thomas J, Chellis J, Miller E, Levinson DF: Genomewide significant linkage to recurrent, early-onset major depressive disorder on chromosome 15q. Am J Hum Genet 2004; 74:1154–1167Google Scholar
23. Levinson DF, Zubenko GS, Crowe RR, DePaulo RJ, Scheftner WS, Weissman MM, Holmans P, Zubenko WN, Boutelle S, Murphy-Eberenz K, MacKinnon D, McInnis MG, Marta DH, Adams P, Sassoon S, Knowles JA, Thomas J, Chellis J: Genetics of recurrent early-onset depression (GenRED): design and preliminary clinical characteristics of a repository sample for genetic linkage studies. Am J Med Genet B Neuropsychiatr Genet 2003; 119B(1):118–130Google Scholar
24. Shi J, Potash JB, Knowles JA, Weissman MM, Coryell W, Scheftner WA, Lawson WB, DePaulo JR Jr, Gejman PV, Sanders AR, Johnson JK, Adams P, Chaudhury S, Jancic D, Evgrafov O, Zvinyatskovskiy A, Ertman N, Gladis M, Neimanas K, Goodell M, Hale N, Ney N, Verma R, Mirel D, Holmans P, Levinson DF: Genome-wide association study of recurrent early-onset major depressive disorder. Mol Psychiatry (in press)Google Scholar
25. Lathrop GM, Lalouel JM: Easy calculations of lod scores and genetic risks on small computers. Am J Hum Genet 1984; 36:460–465Google Scholar
26. O’Connell JR, Weeks DE: Pedcheck: a program for identification of genotype incompatibilities in linkage analysis. Am J Hum Genet 1998; 63:259–266Google Scholar
27. Holmans P, Weissman MM, Zubenko GS, Scheftner WA, Crowe RR, Depaulo JR Jr, Knowles JA, Zubenko WN, Murphy-Eberenz K, Marta DH, Boutelle S, McInnis MG, Adams P, Gladis M, Steele J, Miller EB, Potash JB, Mackinnon DF, Levinson DF: Genetics of recurrent early-onset major depression (GenRED): final genome scan report. Am J Psychiatry 2007; 164:248–258Google Scholar
28. Boehnke M, Cox NJ: Accurate inference of relationships in sib-pair linkage studies. Am J Hum Genet 1997; 61:423–429Google Scholar
29. Kong A, Gudbjartsson DF, Sainz J, Jonsdottir GM, Gudjonsson SA, Richardsson B, Sigurdardottir S, Barnard J, Hallbeck B, Masson G, Shlien A, Palsson ST, Frigge ML, Thorgeirsson TE, Gulcher JR, Stefansson K: A high-resolution recombination map of the human genome. Nat Genet 2002; 31:241–247Google Scholar
30. Smith EN, Bloss CS, Badner JA, Barrett T, Belmonte PL, Berrettini W, Byerley W, Coryell W, Craig D, Edenberg HJ, Eskin E, Foroud T, Gershon E, Greenwood TA, Hipolito H, Koller DL, Lawson WB, Liu C, Lohoff F, McInnis MG, McMahon FJ, Mirel DB, Nievergelt C, Nurnberger J, Nwulia EA, Paschall J, Potash JB, Rice J, Schulze TG, Scheftner W, Panganiban C, Zaitlen N, Zandi PP, Zöllner S, Schork NJ, Kelsoe JR Genome-wide association study of bipolar disorder in European American and African American individuals. Mol Psychiatry 2009; 14:755–763Google Scholar
31. Gudbjartsson DF, Thorvaldsson T, Kong A, Gunnarsson G, Ingolfsdottir A: Allegro version 2. Nat Genet 2005; 37:1015–1016Google Scholar
32. Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC: Plink: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 2007; 81:559–575Google Scholar
33. Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D: Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet 2006; 38:904–90955Google Scholar
34. de Bakker PI, Ferreira MA, Jia X, Neale BM, Raychaudhuri S, Voight BF: Practical aspects of imputation-driven meta-analysis of genome-wide association studies. Hum Mol Genet 17(R2):R122–R128, 2008Google Scholar
35. Stata statistical software: Release 9. College Station, Tex, StataCorp LP, 2005Google Scholar
36. Fallin MD, Lasseter VK, Wolyniec PS, McGrath JA, Nestadt G, Valle D, Liang KY, Pulver AE: Genomewide linkage scan for bipolar-disorder susceptibility loci among Ashkenazi Jewish families. Am J Hum Genet 2004; 75:204–219Google Scholar
37. Kumar HB, Purushottam M, Kubendran S, Gayathri P, Mukherjee O, Murthy AR, Ghosh S, Chandra P, Reddy YC, Benegal V, Brahmachari SK, Jain S: Serotonergic candidate genes and puerperal psychosis: an association study. Psychiatr Genet 2007; 17:253–260Google Scholar
38. Jones I, Middle F, McCandless F, Coyle N, Robertson E, Brockington I, Lendon C, Craddock N: Molecular genetic studies of bipolar disorder and puerperal psychosis at two polymorphisms in the estrogen receptor alpha gene (ESR 1). Am J Med Genet 2000; 96:850–853Google Scholar
39. Robertson E, Jones I, Middle F, Moray J, Craddock N: No association between two polymorphisms at the 5HT2A gene and bipolar affective puerperal psychosis. Acta Psychiatr Scand 2003; 108:387–391Google Scholar
40. Feng J, Zheng J, Bennett WP, Heston LL, Jones IR, Craddock N, Sommer SS: Five missense variants in the amino-terminal domain of the glucocorticoid receptor: no association with puerperal psychosis or schizophrenia. Am J Med Genet 2000; 96:412–417Google Scholar
41. Middle F, Jones I, Robertson E, Lendon C, Craddock N: Tumour necrosis factor alpha and bipolar affective puerperal psychosis. Psychiatr Genet 2000; 10:195–198Google Scholar
42. Carroll JS, Meyer CA, Song J, Li W, Geistlinger TR, Eeckhoute J, Brodsky AS, Keeton EK, Fertuck KC, Hall GF, Wang Q, Bekiranov S, Sementchenko V, Fox EA, Silver PA, Gingeras TR, Liu XS, Brown M: Genome-wide analysis of estrogen receptor binding sites. Nat Genet 2006; 38:1289–1297Google Scholar
43. UCSC Genome Browser, Sestan Lab Human Brain Microarrays track. http://genome.ucsc.eduGoogle Scholar
44. Videbech P, Ravnkilde B: Hippocampal volume and depression: a meta-analysis of MRI studies. Am J Psychiatry 2004; 161:1957–1966Google Scholar
45. Green AD, Galea LA: Adult hippocampal cell proliferation is suppressed with estrogen withdrawal after a hormone-simulated pregnancy. Horm Behav 2008; 54:203–211Google Scholar
46. Métivier R, Gallais R, Tiffoche C, Le Péron C, Jurkowska RZ, Carmouche RP, Ibberson D, Barath P, Demay F, Reid G, Benes V, Jeltsch A, Gannon F, Salbert G: Cyclical DNA methylation of a transcriptionally active promoter. Nature 2008; 452:45–50Google Scholar

