
Evidence for an Interaction Between Familial Liability and Prenatal Exposure to Infection in the Causation of Schizophrenia
Abstract
Objective: The authors sought to determine whether prenatal exposure to infection and a positive family history of psychotic disorders interact synergistically to increase the risk of later developing schizophrenia. Method: The authors linked two national registers, the Medical Birth Register and the Finnish Population Register, to identify all women in Helsinki who received hospital treatment during pregnancy for an upper urinary tract infection (N=9,596) between 1947 and 1990. The Finnish Hospital Discharge Register was used to ascertain psychiatric outcomes in adulthood of offspring exposed to infection prenatally. Family history of psychotic disorders was determined by linking the Hospital Discharge Register and the Population Register. The authors used an additive statistical interaction model to calculate the amount of biological synergism between positive family history and prenatal exposure to infection. Results: Prenatal exposure to infection did not significantly increase the risk of schizophrenia. However, the effect of prenatal exposure to pyelonephritis was five times greater in those who had a family history of psychosis compared to those who did not. The synergy analysis suggested that an estimated 38%–46% of the offspring who developed schizophrenia and had both prenatal exposure to infection and a positive family history of psychotic disorders did so as a result of the synergistic action of both risk factors. Conclusions: These findings support a mechanism of gene-environment interaction in the causation of schizophrenia.
The search for risk factors for schizophrenia is taking us ever further back in development (1) . Prenatal exposure to infection has been the subject of particular interest since Mednick et al. (2) first reported an increased risk of schizophrenia among individuals exposed prenatally to the 1957 influenza epidemic in Helsinki. Most subsequent studies examining this issue have found a positive association, but there have also been some negative findings (see references 3 – 6 for reviews). Early studies pointed to the importance of exposure to infection during the second trimester of fetal development, but recent work places the window of vulnerability in the first trimester (7 – 9) . A wide variety of infections, such as influenza, herpes, polio, rubella, toxoplasmosis, and respiratory infections, have been implicated, indicating that some pathogenic mechanism common to many infections, such as fever or production of cytokines, may be in operation (8 , 10) .
The effect sizes reported for the association between prenatal exposure to infection and later schizophrenia have generally been in the region of a 1.5- to 2-fold increase in risk (4 , 11) . While it is possible that these small effect sizes represent random noise and that no real association exists, recent studies using prenatal maternal serum samples have found that individuals exposed prenatally to rubella had a 10- to 20-fold increase in risk of adult schizophrenia (12) and that those exposed to influenza during the first trimester of gestation had a sevenfold increase in risk (8) . The presence of inconsistencies among findings across studies suggests that prenatal exposure to infection may be interacting with another risk factor for schizophrenia, such as genetic risk, to produce its effect (13) .
The importance of considering genetic liability to psychosis when examining the effect of prenatal environmental exposures has been noted by numerous authors (1 – 3 , 8 , 14 – 16) . However, previous studies either have lacked the information needed to examine such effects or have had insufficient statistical power to do so (2 , 8 , 17 – 20) . Some investigators have included parental psychiatric history as a potential confounder in the analysis (21 , 22) , but this approach is not adequate to explore the possible synergistic effects of genetic liability to psychosis and an adverse environmental exposure (23) .
In this study we examined the effect of prenatal exposure to pyelonephritis, an infection of the upper urinary tract, on the subsequent development of schizophrenia in a large Finnish cohort. Pyelonephritis is particularly common in pregnancy, occurring in 1%–2% of pregnant women. Because most pregnant women with this infection are admitted to the hospital (24) , hospital records allowed us to accurately time the exposure to infection during gestation. Record linkage between population and hospital discharge registers gave us the information needed to model the synergistic effects of prenatal infection exposure and family history of psychosis.
We had three hypotheses:
1. Hospital-treated acute pyelonephritis during pregnancy is associated with an increased risk of schizophrenia and other psychotic disorders in exposed offspring.
2. The effects of prenatal exposure to infection are dependent on the timing of the exposure during pregnancy. Based on evidence from recent animal work and archived maternal serum studies, we expected that exposure during the first trimester would have the biggest impact on risk (8 , 9) .
3. There is a synergistic relationship between prenatal exposure to infection and family history of psychotic disorders in increasing the risk of psychotic disorders in adulthood.
Method
All live-born individuals in Finland are assigned a unique personal identification number at birth. This number is used in all registers, which ensures accurate linkage of information between registers. For this study, three national registers were linked: the Finnish Hospital Discharge Register, the Medical Birth Register, and the Finnish Population Register. These registers are maintained in computerized databases by the National Research and Development Center for Health and Welfare.
The base study population comprised all individuals born in Helsinki between 1947 and 1990. From this study population, using record linkage between the Medical Birth Register and the Population Register, we identified all individuals whose mothers were hospitalized for acute pyelonephritis in pregnancy, that is, while members of the study population were in gestation. The Medical Birth Register had information on the exact week of pregnancy during which the women were treated in the hospital for pyelonephritis. Siblings of these individuals who were not exposed in utero to maternal pyelonephritis were taken as the comparison group. Siblings were identified through linkage with the Population Register.
Psychiatric outcomes in adulthood and family history of psychosis were determined by linkage to the Finnish Hospital Discharge Register, which covers all public and private hospitals in Finland. Psychiatric diagnoses were ascertained for exposed cases, the sibling comparison group, and parents. Psychiatric diagnoses are recorded in the register using ICD codes. Diagnoses before 1987 were made using ICD-8, diagnoses between 1987 and 1995 were made using ICD-9, and since 1995 diagnoses have been made using ICD-10. In this study we defined “schizophrenia” as a 295 diagnosis in ICD-8 and ICD-9 or an F20 diagnosis in ICD-10. The following diagnoses were used for “broadly defined psychotic disorders”: ICD-8/9 codes 295 (schizophrenia), 296 (affective psychoses), 297 (delusional syndrome), 298 (reactive psychoses), and 299 (childhood origin psychoses) and ICD-10 codes F20 (schizophrenia), F21 (schizotypal disorder), F22 (delusional syndrome), F23 (psychotic episode), F24 (induced delusional syndrome), F25 (schizoaffective syndrome), F28 (other psychoses), F29 (psychosis not otherwise specified), F30.2 (mania with psychotic symptoms), F31.2 (bipolar affective disorder with psychotic symptoms), F31.5 (bipolar affective disorder, depression with psychotic symptoms), F32.3 (severe depressive episode with psychotic symptoms), and F33.3 (recurrent depressive disorder with psychotic symptoms). A positive family history of psychosis was defined as having a parent or sibling with a diagnosis of broadly defined psychotic disorder on the Finnish Hospital Discharge Register.
Statistical Analyses
Statistical analyses were conducted using Stata statistical software, version 8.0 (25). A logistic regression model was used to calculate odds ratios and 95% confidence intervals (CIs) with schizophrenia or broadly defined psychotic disorder as the response variable and in utero exposure to infection as the exposure variable. Standard errors were corrected for clustering within families using the ROBUST function in Stata. The effect of exposure to infection at different times during pregnancy on the later development of psychotic disorders in the offspring was examined by analyzing the data according to trimester. Trimester 1 was defined as weeks 1–12 of pregnancy, trimester 2 as weeks 12–28, and trimester 3 as weeks 28–40.
To examine the synergistic effect of exposure to infection and genetic liability to psychosis, we stratified the data on the basis of family history of psychotic disorder and examined the effect of prenatal infection within history-positive and history-negative strata. We calculated the statistical additive interaction between family history and infection exposure (i.e., as a risk difference rather than a risk ratio) using risk difference regression. The BINREG procedure in Stata, which fits generalized linear models for the binomial family estimating risk differences, was used. The statistical significance of the interactions was assessed using the Wald test (26) . We then estimated the amount of biological synergism between prenatal infection exposure and family history of psychotic disorder using methods developed by Darroch (27) and previously used by van Os et al. (23 , 28) (see the data supplement that is included with the online edition of this article).
Results
Prenatal Exposure to Infection and Later Schizophrenia
From the national registers we identified 9,596 individuals born between 1947 and 1990 who were in gestation when their mothers were hospitalized for pyelonephritis. The comparison group comprised 13,808 siblings of these individuals, who were also born between 1947 and 1990 and who were not exposed to maternal pyelonephritis while in utero. From record linkage we ascertained that 6.4% of the sample had a positive family history of psychotic disorder ( Table 1 ).
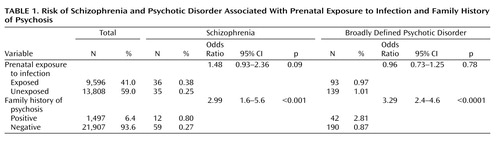
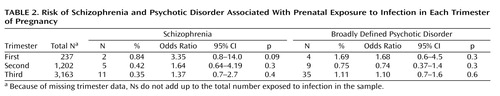
Table 1 provides the odds ratios for schizophrenia and broadly defined psychotic disorder in case and comparison groups in relation to prenatal exposure to infection and family history. Exposure to infection did not significantly increase the risk of developing either schizophrenia or broadly defined psychotic disorder. However, in a result that approached statistical significance, the risk of developing schizophrenia was almost 50% higher in the exposed group compared to the unexposed group. Table 2 shows that the timing of the infection during gestation did not significantly affect the risk of schizophrenia, but there was some evidence approaching statistical significance that first-trimester exposure to infection increased the risk of later schizophrenia but not of broadly defined psychotic disorder. There was no association between family history of psychotic disorder and in utero exposure to infection. Therefore, our results were not confounded by any increased tendency for mothers with psychotic illness to develop infection during pregnancy compared to those without psychotic illness.
Interaction Between Prenatal Exposure to Infection and Family History of Psychosis
The results of the interaction analysis are presented in Table 3 . The effect of prenatal exposure to pyelonephritis was significantly greater in those who had a family history of psychotic disorder compared to those who did not. Stratified analyses revealed that the difference in risk for schizophrenia (i.e., risk difference) between those exposed and those not exposed to prenatal infection in the genetically vulnerable group was five times larger than the risk difference in those who were not genetically vulnerable (interaction χ 2 =7.14, df=1, p=0.02).

We calculated the amount of synergism between the two risk factors. The risk percentages for each risk stratum were as follows: no infection/no family history, 0.23% (30/12,610); infection only, 0.32% (29/8,956); family history only, 0.58% (5/857); and infection/family history, 1.09% (7/640). These risk percentages were entered into a standard formula for calculating the amount of biological synergism between two risk factors (23 , 27 , 28 ; see the online data supplement). This analysis suggested that 38%–46% of the individuals who developed schizophrenia in our sample did so as a result of the synergistic action of prenatal exposure to infection and positive family history.
Discussion
To our knowledge, this is the first report in the schizophrenia literature showing a gene-environment synergistic effect between prenatal exposure to infection and familial liability to psychosis. We found an additive effect of having a positive family history such that, when coupled with prenatal exposure to infection, the risk is significantly increased relative to those who were exposed to infection but are not at increased familial risk. In those who are genetically vulnerable to psychotic disorder, the risk difference between those exposed and those unexposed to infection was five times larger than the risk difference between the exposed and the unexposed among those without genetic vulnerability. As noted by van Os et al. (23) , this type of additive analysis is necessary for detecting the synergistic effects between genetic and environmental risk factors for mental illness.
The effect size found here for prenatal exposure to infection is modest but is in the range of what has previously been found (1) and is larger than the effect size found for most putative susceptibility genes (29) . The analysis of the timing of exposure to infection during pregnancy suggests the possible importance of first-trimester exposure, but because only two patients were exposed in the first trimester, no conclusions can be drawn. Although many previous studies have found the strongest effects for the second trimester of pregnancy (4) , these studies were based on ecological data and suffered from a lack of exact information in relation to the definition of exposure. In line with our findings, recent animal research (9) and work using archived maternal serum (8) has found that exposure to infection during the first trimester or very early in the second trimester of pregnancy, compared to later periods in pregnancy, confers the greatest risk of developing schizophrenia. A recent review of this issue (30) supports the hypothesis that infection-associated immunological events in early fetal life have a stronger neurodevelopmental impact than such events later in pregnancy.
Strengths and Limitations
Our study has a number of strengths. First, the sample size was large, providing adequate statistical power to examine statistical additive effects. Second, the use of siblings as the comparison group meant that exposed and unexposed individuals were likely to have experienced similar environmental and social influences during childhood that might otherwise represent unknown confounders. The use of a sibling comparison group also controlled for a difference in genetic risk of psychotic disorder between our groups. The majority of previous studies lacked information on genetic risk in their samples; it is possible that these studies had more individuals at increased genetic risk of psychotic disorders in their exposed groups, thus inflating the effect size found for infection exposure. Third, the use of hospital-treated pyelonephritis to establish the infection group gave us exact information on the timing of exposure to infection during pregnancy.
The study also had a number of limitations. Although our sample was large, we had a small number of cases of schizophrenia in each group when the sample was stratified by infection exposure and family history, and we were missing trimester data on a significant proportion of our infection-exposed individuals. Therefore, we did not have sufficient statistical power to investigate whether there was an interaction between prenatal infection exposure and family history of psychotic disorder within each trimester. Given the trend in the data for a first-trimester effect, this would be an interesting analysis in a study with a larger sample. Another potential limitation is that nearly all women with pyelonephritis are treated with antibiotics. We have no data on the type of antibiotics used. Therefore, a confounding effect of medication cannot be ruled out. It is not inconceivable that the gene-environment interaction found here is actually a gene-medication interaction.
Possible Mechanisms
Despite much speculation, the mechanism by which prenatal infection increases the risk of schizophrenia has not yet been elucidated. An indirect effect of infection on fetal brain development via maternal immunoglobulin G antibodies elicited by the infection or via maternal cytokines would seem to be the most plausible pathogenic mechanism (6 , 31) . In support of this hypothesis, a positive association between elevated maternal levels of the cytokine interleukin-8 (IL-8) during pregnancy and an increased risk of schizophrenia spectrum disorders in the offspring has been reported (32) , and IL-8 has been found to be elevated in children with acute pyelonephritis (33) . The possibility of a direct effect, of either the infectious organism itself or the associated high fever, should be considered. Escherichia coli is the most common bacterium causing pyelonephritis and is involved in 65%–80% of cases. However, since most urinary tract infections are not accompanied by bloodstream infection, even in pregnancy a direct effect of the infection is unlikely (34 – 36) . A direct effect of hyperthermia is also a possibility as pyelonephritis is clinically associated with high fever (24) . Hyperthermia during pregnancy has been shown to significantly increase the risk of a range of negative pregnancy outcomes, including major structural malformations, such as neural tube defects (37 – 40) .
Gene-Environment Interaction in Schizophrenia
Gene-environment interactions have recently been demonstrated in psychiatric illnesses such as ADHD, depression, posttraumatic stress disorder, and conduct disorder (13 , 41) . In schizophrenia, it has been shown that offspring at high genetic risk for schizophrenia are particularly sensitive to the adverse effects of a negative family rearing environment (42) , that the link between early cannabis use and later psychosis is strongest in a certain genetic subgroup (43) , and that the effect of urbanicity is mediated by genetic liability to psychosis (23 , 28) . The urbanicity studies, which carried out the kind of additive interaction analysis that we used in this study, found that 60%–70% of those who developed schizophrenia and were exposed to both risk factors examined did so as a result of the synergistic action between urban dwelling and familial liability to psychosis (23) . We found that up to half of the cases of schizophrenia in our sample who were exposed to both risk factors could be attributed to the synergistic action of prenatal infection exposure and familial liability to psychosis.
Research using gene-environment strategies is providing robust evidence of the power of looking at environmental exposures in the context of increased genetic risk for disorders with a complex etiology, such as schizophrenia. Infectious exposures are a good candidate for environmental pathogens for use in such analyses for several reasons. First, variability in response to exposure has been observed, indicating that the outcome is dependent on at least one other variable. Second, a number of studies have provided evidence that infection is causally related to schizophrenia, indicating that the association is not wholly genetically mediated. And third, the maternal immune response to infection represents a neurobiologically plausible pathway along which infection may increase the risk for schizophrenia (41) . Until the exact mechanism by which prenatal exposure to infection increases risk of psychosis is elucidated, studies such as this, using proxy measures of environmental risk and genetic liability, will remain useful.
1. Cannon M, Clarke MC: Risk for schizophrenia: broadening the concepts, pushing back the boundaries. Schizophr Res 2005; 79:5–13Google Scholar
2. Mednick SA, Machon RA, Huttunen MO, Bonett D: Adult schizophrenia following prenatal exposure to an influenza epidemic. Arch Gen Psychiatry 1988; 45:189–192Google Scholar
3. Brown AS: Prenatal infection as a risk factor for schizophrenia. Schizophr Bull 2006; 32:200–202Google Scholar
4. Cannon M, Kendell R, Susser E, Jones P: Prenatal and perinatal risk factors for schizophrenia, in The Epidemiology of Schizophrenia. Edited by Murray RM, Jones PB, Susser E, van Os J, Cannon M. Cambridge, England, Cambridge University Press, 2002, pp 74–99Google Scholar
5. Watson JB, Mednick SA, Huttunen M, Wang X: Prenatal teratogens and the development of adult mental illness. Dev Psychopathol 1999; 11:457–466Google Scholar
6. Wright P, Takei N, Murray RM, Sham PC: Seasonality, prenatal influenza exposure, and schizophrenia, in Prenatal Exposures in Schizophrenia. Edited by Susser ES, Brown AS, Gorman JM. Washington, DC, American Psychiatric Press, 1999, pp 89–112Google Scholar
7. Brown AS, Cohen P, Greenwald S, Susser E: Nonaffective psychosis after prenatal exposure to rubella. Am J Psychiatry 2000; 157:438–443Google Scholar
8. Brown AS, Begg MD, Gravenstein S, Schaefer CA, Wyatt RJ, Bresnahan M, Babulas VP, Susser ES: Serologic evidence of prenatal influenza in the etiology of schizophrenia. Arch Gen Psychiatry 2004; 61:774–780Google Scholar
9. Fatemi SH, Pearce DA, Brooks AI, Sidwell RW: Prenatal viral infection in mouse causes differential expression of genes in brains of mouse progeny: a potential animal model for schizophrenia and autism. Synapse 2005; 57:91–99Google Scholar
10. Thomas KL, Huttunen M: Neural migration, proinflammatory interleukins, and periventricular leukomalacia: involvement in schizophrenia. Trends Neurosci 1999; 22:389–390Google Scholar
11. Clarke MC, Harley M, Cannon M: The role of obstetric events in schizophrenia. Schizophr Bull 2006; 32:3–8Google Scholar
12. Brown AS, Cohen P, Harkavy-Friedman J, Babulas V, Malaspina D, Gorman JM, Susser ES: Prenatal rubella, premorbid abnormalities, and adult schizophrenia. Biol Psychiatry 2001; 49:473–486Google Scholar
13. Caspi A, Moffitt TE: Gene-environment interactions in psychiatry: joining forces with neuroscience. Nat Rev Neurosci 2006; 7:583–590Google Scholar
14. Huttunen MO, Machon RA, Mednick SA: Prenatal factors in the pathogenesis of schizophrenia. Br J Psychiatry Suppl 1994; 23:15–19Google Scholar
15. Isohanni M, Isohanni I, Koponen H, Koskinen J, Laine P, Lauronen E, Miettunen J, Mäki P, Riala K, Räsänen S, Saari K, Tienari P, Veijola J, Murray G: Developmental precursors of psychosis. Curr Psychiatry Rep 2004; 6:168–175Google Scholar
16. van OS J, Sham P: Gene-environment correlation and interaction in schizophrenia, in The Epidemiology of Schizophrenia. Edited by Murray RM, Jones PB, Susser E, van Os J, Cannon M. Cambridge, England, Cambridge University Press, 2003, pp 235–253Google Scholar
17. Sørensen HJ, Mortensen EL, Reinisch JM, Mednick SA: Association between prenatal exposure to analgesics and risk of schizophrenia. Br J Psychiatry 2004; 185:366–371Google Scholar
18. O’Callaghan E, Sham P, Takei N, Glover G, Murray RM: Schizophrenia after prenatal exposure to 1957 A2 influenza epidemic. Lancet 1991; 337:1248–1250Google Scholar
19. Takei N, Sham P, O’Callaghan E, Murray GK, Glover G, Murray RM: Prenatal exposure to influenza and the development of schizophrenia: is the effect confined to females? Am J Psychiatry 1994; 151:117–119Google Scholar
20. Wright P, Takei N, Rifkin L, Murray RM: Maternal influenza, obstetric complications, and schizophrenia. Am J Psychiatry 1995; 152:1714–1720Google Scholar
21. Westergaard T, Mortensen PB, Pedersen CB, Wohlfahrt J, Melbye M: Exposure to prenatal and childhood infections and the risk of schizophrenia: suggestions from a study of sibship characteristics and influenza prevalence. Arch Gen Psychiatry 1999; 56:993–998Google Scholar
22. Mortensen PB, Nørgaard-Pedersen B, Waltoft BL, Sørensen TL, Hougaard D, Yolken RH: Early infections of Toxoplasma gondii and the later development of schizophrenia. Schizophr Bull 2007; 33:741–744 Google Scholar
23. van Os J, Hanssen M, Bak M, Bijl RV, Vollebergh W: Do urbanicity and familial liability coparticipate in causing psychosis? Am J Psychiatry 2003; 160:477–482Google Scholar
24. Ramakrishnan K, Scheid DC: Diagnosis and management of acute pyelonephritis in adults. Am Fam Physician 2005; 71:933–942Google Scholar
25. StataCorp: Stata, release 8.0. College Station, Tex, StataCorp, 2005Google Scholar
26. Clayton D, Hills M: Wald tests, in Statistical Models in Epidemiology. Edited by Clayton D, Hills M. Oxford, England, Oxford Science Publications, 1993, pp 101–102Google Scholar
27. Darroch J: Biologic synergism and parallelism. Am J Epidemiol 1997; 145:661–668Google Scholar
28. van Os J, Pedersen CB, Mortensen PB: Confirmation of synergy between urbanicity and familial liability in the causation of psychosis. Am J Psychiatry 2004; 161:2312–2314Google Scholar
29. Kendler KS: “A Gene for…”: the nature of gene action in psychiatric disorders. Am J Psychiatry 2005; 162:1243–1252Google Scholar
30. Meyer U, Yee BK, Feldon J: The neurodevelopmental impact of prenatal infections at different times of pregnancy: the earlier the worse? Neuroscientist 2007; 13:241–256Google Scholar
31. Gilmore JH, Jarskog LF: Exposure to infection and brain development: cytokines in the pathogenesis of schizophrenia. Schizophr Res 1997; 24:365–367Google Scholar
32. Brown AS, Hooton J, Schaefer CA, Zhang H, Petkova E, Babulas V, Perrin M, Gorman JM, Susser ES: Elevated maternal interleukin-8 levels and risk of schizophrenia in adult offspring. Am J Psychiatry 2004; 161:889–895Google Scholar
33. Sheu JN, Chen MC, Lue KH, Cheng SL, Lee IC, Chen SM, Tsay GJ: Serum and urine levels of interleukin-6 and interleukin-8 in children with acute pyelonephritis. Cytokine 2006; 36:276–282Google Scholar
34. Peterson J, Kaul S, Khashab M, Fisher A, Kahn JB: Identification and pretherapy susceptibility of pathogens in patients with complicated urinary tract infection or acute pyelonephritis enrolled in a clinical study in the United States from November 2004 through April 2006. Clinical Therapeutics 2007; 29:2215–2221Google Scholar
35. Hill JB, Sheffield JS, McIntire DD, Wendel GD Jr: Acute pyelonephritis in pregnancy. Obstet Gynecol 2005; 105:18–23Google Scholar
36. Ronald A: The etiology of urinary tract infection: traditional and emerging pathogens. Am J Med 2002; 113(suppl 1A):14S–19SGoogle Scholar
37. Milunsky A, Ulcickas M, Rothman KJ, Willett W, Jick SS, Jick H: Maternal heat exposure and neural tube defects. JAMA 1992; 268:882–885Google Scholar
38. Little BB, Ghali FE, Snell LM, Knoll KA, Johnston W, Gilstrap LC 3rd: Is hyperthermia teratogenic in the human? Am J Perinatol 1991; 8:185–189Google Scholar
39. Chambers CD, Johnson KA, Dick LM, Felix RJ, Jones KL: Maternal fever and birth outcome: a prospective study. Teratology 1998; 58:251–257Google Scholar
40. Suarez L, Felkner M, Hendricks K: The effect of fever, febrile illnesses, and heat exposures on the risk of neural tube defects in a Texas-Mexico border population. Birth Defects Res A Clin Mol Teratol 2004; 70:815–819Google Scholar
41. Moffitt TE, Caspi A, Rutter M: Strategy for investigating interactions between measured genes and measured environments. Arch Gen Psychiatry 2005; 62:473–481Google Scholar
42. Tienari P, Wynne LC, Sorri A, Lahti I, Läksy K, Moring J, Naarala M, Nieminen P, Wahlberg KE: Genotype-environment interaction in schizophrenia-spectrum disorder: long-term follow-up study of Finnish adoptees. Br J Psychiatry 2004; 184:216–222Google Scholar
43. Caspi A, Moffitt TE, Cannon M, McClay J, Murray R, Harrington H, Taylor A, Arseneault L, Williams B, Braithwaite A, Poulton R, Craig IW: Moderation of the effect of adolescent-onset cannabis use on adult psychosis by a functional polymorphism in the catechol- O -methyltransferase gene: longitudinal evidence of a gene environment interaction. Biol Psychiatry 2005; 57:1117–1127 Google Scholar

