
Genetic Markers of Suicidal Ideation Emerging During Citalopram Treatment of Major Depression
Abstract
Objective: Suicidal ideation is an uncommon symptom than can emerge during antidepressant treatment. The biological basis of treatment-emergent suicidal ideation is unknown. Genetic markers may shed light on the causes of treatment-emergent suicidal ideation and help identify individuals at high risk who may benefit from closer monitoring, alternative treatments, or specialty care. Method: A clinically representative cohort of outpatients with major depressive disorder who enrolled in the Sequenced Treatment Alternatives to Relieve Depression (STAR*D) trial were treated with citalopram under a standard protocol for up to 14 weeks. DNA samples from 1,915 participants were genotyped for 768 single-nucleotide polymorphisms in 68 candidate genes. Allele and genotype frequencies were compared between the 120 participants who developed treatment-emergent suicidal ideation and those who did not. Results: Two markers were significantly associated with treatment-emergent suicidal ideation in this sample (marker rs4825476, p=0.0000784, odds ratio=1.94; permutation p=0.01; marker rs2518224, p=0.0000243, odds ratio=8.23; permutation p=0.003). These markers reside within the genes GRIA3 and GRIK2, respectively, both of which encode ionotropic glutamate receptors. Conclusions: Markers within GRIK2 and GRIA3 were associated with treatment-emergent suicidal ideation during citalopram therapy. If replicated, these findings may shed light on the biological basis of this potentially dangerous adverse event and help identify patients at increased risk.
Since the introduction of selective serotonin reuptake inhibitor (SSRI) antidepressants in the 1980s, there has been controversy over whether they can trigger suicidal thoughts or behavior (1 , 2) . In 1991, the Psychopharmacologic Drugs Advisory Committee of the U.S. Food and Drug Administration (FDA) found no clear evidence of increased suicide risk associated with fluoxetine (3) . In 2003, however, the U.K. Medicines and Healthcare Products Regulatory Agency concluded that paroxetine, citalopram, and other SSRIs were contraindicated in youths because of an increased risk of treatment-emergent suicidal ideation (4) . Soon after that, a scrupulous analysis by the FDA and an independent group at Columbia University ultimately resulted in an FDA-mandated black box warning highlighting the potential for suicidal ideation in youths treated with SSRIs. The warning was later extended to all antidepressants (5) . Since the black box warning was issued, the use of SSRIs in children has decreased (6) and concern about treatment-emergent suicidal ideation has extended to adults.
Treatment-emergent suicidal ideation and behavior are infrequent; the average incidence is 4% for antidepressants and 2% for placebo (7) . The highest risk seems to be within a few weeks after initiation of treatment or dose adjustment (8 , 9) . It is not clear whether treatment-emergent suicidal ideation heralds actual suicidal behavior in children or adults. Of the 4,400 pediatric subjects who participated in the clinical trials of SSRIs analyzed by the FDA, no completed suicides were reported (7) .
The Sequenced Treatment Alternatives to Relieve Depression (STAR*D) trial, the largest prospective treatment trial for major depressive disorder, provides a unique opportunity to ascertain treatment-emergent suicidal ideation prospectively in a large cohort of patients treated with the SSRI citalopram and to test whether specific genetic markers can identify patients who have an increased risk of developing this uncommon adverse event. Here we report an initial screen with 768 markers in 68 candidate genes. The results suggest that genetic markers may be able to identify some people at increased risk of treatment-emergent suicidal ideation. If replicated, these findings could have implications for the clinical management of major depression with SSRIs.
Method
Sample
Ascertainment and evaluation for the STAR*D study have been detailed elsewhere (10 , 11) . Briefly, investigators implemented a standard study protocol at 18 primary care and 23 psychiatric care settings across the United States. Participants provided written informed consent for both the treatment study and for DNA collection, although participation in DNA collection was optional. The genetic protocol was initiated approximately 12 months after treatment study initiation. Outpatients 18–75 years of age who had an initial score ≥14 on the 17-item Hamilton Depression Rating Scale (HAM-D; 12 , 13) and who met DSM-IV (14) criteria for nonpsychotic major depressive disorder were eligible. Patients with bipolar or psychotic disorders were excluded, as were those with a primary diagnosis of obsessive-compulsive disorder or an eating disorder, a general medical condition in which study medications were contraindicated, substance dependence requiring inpatient detoxification, or clear nonresponse or intolerance to any protocol antidepressant during the current episode. Patients who were pregnant or breastfeeding were also excluded.
At the first treatment step (Level 1), all participants received citalopram, typically starting at 20 mg/day, with dose increases following recommended procedures (up to 40 mg/day by week 4 and 60 mg/day by week 6) (15) . The protocol required an adequate dose of citalopram for a sufficient time to ensure that those whose symptoms did not improve were most likely unresponsive to the medication (15) . No concomitant psychotropic medications were allowed aside from benzodiazepines, hypnotics, or trazodone up to 200 mg/day for sleep, if needed. The sample characteristics have been presented elsewhere (15 , 16) .
DNA samples were collected from 1,953 participants. A sample of 20 ml of whole blood was collected and shipped to the Rutgers Cell and DNA Repository, where lymphocytes were extracted and cryopreserved by standard methods. DNA was extracted by standard methods (16) . Samples were arrayed robotically, then gender-verified with a set of three X-linked and two Y-linked markers. A CONSORT (Consolidated Standards of Reporting Trials) diagram of the study sample is shown in Figure 1 .
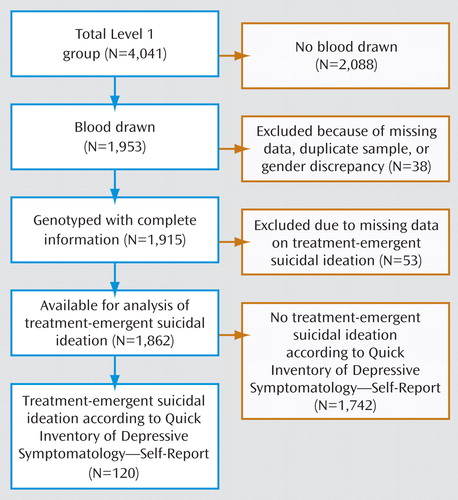
Participants who consented to DNA collection were similar to those in the full sample but differed in some variables (see reference 16 for details). These differences cannot affect the genetic association results, which derive from comparisons among the genotyped subjects, but they do limit generalizability. Those who provided DNA did not differ in the frequency of treatment-emergent suicidal ideation in this sample ( Table 1 ).
Phenotype Definition
Consistent with previous definitions of treatment-emergent suicidal ideation, we used the “thoughts of death or suicide” question (item 12) from the 16-item Quick Inventory of Depressive Symptomatology—Self-Report (QIDS-SR), a reliable and well-validated measure of symptom severity that has been shown to correlate well with the HAM-D (17 – 21) . The QIDS-SR was chosen over the clinician-rated version (the QIDS-C) because suicidal ideation is a subjective phenomenon, and we wished to avoid any clinician bias. The QIDS-SR has been shown to successfully substitute for the QIDS-C and the HAM-D (21) . Secondary testing with the QIDS-C was carried out for markers significantly associated with treatment-emergent suicidal ideation by the QIDS-SR, solely to test the robustness of the findings. The primary outcome phenotype was decided prior to any data analysis.
The QIDS-SR was administered at baseline and at each of the protocol-recommended clinic visits around weeks 2, 4, 6, 9, and 12. Possible responses to item 12 include: “I do not think of suicide or death” (coded 0), “I feel that life is empty or wonder if it is worth living” (coded 1), “I think of suicide or death several times a week for several minutes” (coded 2), and “I think of suicide or death several times a day in some detail, or I have made specific plans for suicide, or have actually tried to take my life” (coded 3). Participants who scored 0 on this item before citalopram treatment and 1, 2, or 3 at least once during treatment were defined as having treatment-emergent suicidal ideation (N=120 cases).
The control group (N=1,742) consisted of all participants who scored 0 on item 12 of the QIDS-SR during up to 12 weeks of citalopram treatment. This included participants who denied any suicidal ideation at the initial and subsequent visits (N=765) and participants who acknowledged suicidal ideation at the initial visit before the start of treatment (N=977). Participants for whom suicidal ideation data were missing at the initial visit or at all subsequent visits were excluded (N=53). We chose this set of control subjects in order to avoid detecting markers that might be associated with general suicidal thoughts unrelated to treatment.
Population Structure
We evaluated the possibility of population structure leading to inflated association results in three ways. First, we used self-reported race as a forced covariate in the logistic regression analysis. We have previously shown that self-reported race corresponds well in this sample to population assignment on the basis of multilocus allele frequencies (15) . Second, we stratified the sample by self-reported race, then investigated the possibility of cryptic structure within the largest subset, those self-described as white (N=1,473). Within this subset, we used the Kolmogorov-Smirnov test to assess whether the p values of the association test were consistent with a uniform distribution under the assumption of one population, as recommended by Pritchard et al. (22) . Finally, we used STRUCTURE and STRAT (23) to evaluate the worst-case scenario of two cryptic populations within the white subset by assessing the multilocus chi-square value in a set of 344 unlinked single-nucleotide polymorphisms selected from the total data set without regard to the association results. We ran STRUCTURE for 20,000 burn-in steps followed by 20,000 replications.
Candidate Genes
Sixty-eight genes were chosen for study from among a larger list of plausible candidates, which has been detailed elsewhere (16) . Genes were selected to sample five broad signaling pathways of potential importance in antidepressant effects: serotonin (20 genes), glutamate (16 genes), dopamine (three genes), norepinephrine (four genes), and neurotrophins (four genes), along with selected genes in other pathways (21 genes).
Selection of Single-Nucleotide Polymorphism Markers
A total of 768 single-nucleotide polymorphisms were selected to sample common variation, as detailed elsewhere (16) . Briefly, genotype data spanning the coding region and up to 2 kilobases of flanking sequence were downloaded from HapMap database (www.hapmap.org, accessed Nov. 2004). The program “LDSelect” (24) was used to choose an optimal set of available single-nucleotide polymorphisms to genotype, at an r 2 threshold of >0.8, excluding single-nucleotide polymorphisms with a minor allele frequency <7.5%. (The complete list of single-nucleotide polymorphisms genotyped is available from the first author upon request.)
Genotyping Methods
Samples were shipped to Illumina, Inc. (San Diego), where they were genotyped on their BeadArray platform, a highly accurate assay (25) . The genotyping success rate was 99.9%, and 99.73% of samples were successfully genotyped, including 35,052 blind duplicate genotypes, all of which matched exactly.
Power analysis was performed with the Genetic Power Calculator ( 26 ; http://pngu.mgh.harvard.edu/~purcell/gpc/). The number of cases was set at the observed 120, with a control-to-case ratio at the observed value of 15. The high-risk allele frequency was set at 0.3. The trait prevalence was set at the observed value of 0.062. Marker-disease allele linkage disequilibrium (D′) was set to 0.81, equal to the median D′ value observed in the actual data (16) . In an allele-wise test, power was greater than 80% to detect association at the p=0.001 level with a variant conferring a heterozygote relative risk of 1.8. Power dropped to 60% at p=0.0001 but exceeded 94% at p=0.01. In a genotypic association test, with a heterozygote relative risk of 1 and a homozygous relative risk of 4, power was 68% at p=0.001.
Statistical Methods
To improve the sensitivity of our initial analyses, we performed both allelic and genotypic association tests on all markers. Allelic tests are most powerful for alleles that confer risk in a codominant or dominant fashion, while genotypic tests are more powerful when a recessive model applies (27) . Allelic comparisons were performed with Cocaphase in the UNPHASED package (28) , which estimates a likelihood-based test of association under the null hypothesis of all odds ratios being equal to one. Genotypic comparisons were carried out using a Pearson chi-square test on a 2×3 contingency table. To properly account for X-linked markers, these were analyzed separately. The Hardy-Weinberg equilibrium for the chosen markers was calculated using PEDSTATS (29) . Odds ratios for the genotypic test are based on a comparison between the two groups of homozygotes.
Experimentwise p values that correct for the number of single-nucleotide polymorphisms tested were estimated by permuting case-control labels 10,000 times. Allelic p values were permutation-tested in an allelic test; genotypic p values were permutation-tested in a genotypic test. Since autosomal and X-linked markers require different analytic procedures, permutation tests were run separately for autosomal and X-linked markers. The total number of p values less than or equal to the lowest p value observed in the actual data were tallied across the autosomal and X-linked permutation results and then divided by 10,000 to yield the experimentwise p value.
Single-nucleotide polymorphisms that passed the initial tests with an experimentwise p<0.05 were studied further. Tests of association included a logistic regression model calculated with SAS 9.1.3 Enterprise Guide 3.0 (SAS Institute, Cary, N.C.), with a nominal dependent variable for treatment-emergent suicidal ideation. X-linked markers were analyzed in males and females separately. Single-marker tests were carried out under codominant, dominant, and recessive models. Models were compared with the likelihood ratio test. The best-fitting model was used for the multimarker analyses. The reference model was based on the single-nucleotide polymorphisms with the largest identified odds ratios in the single-marker models. The remaining covariates were added in a stepwise fashion in descending order of effect size, as recommended by Cordell and Clayton (30) . The –2 log-likelihood was used to assess the improvement of fit as each variable was removed from the model. The Hosmer-Lemeshow test was used to test for final model fit. For the X-linked marker in GRIA3, these analyses were carried out separately in males and females because there is no method in SAS Enterprise Guide that corrects for X-linked markers in a regression analysis.
Case and control subjects were characterized clinically using univariate tests. Chi-square tests were used to assess differences between groups for nominal and ordinal variables, and t tests for continuous variables.
Results
Descriptive Variables
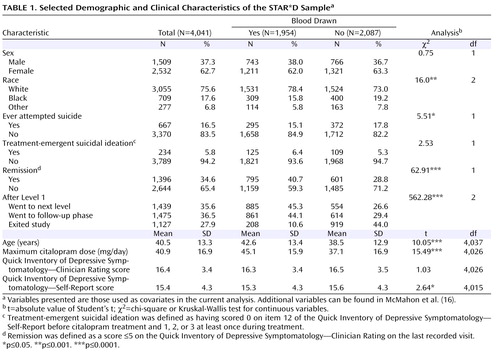
The demographic and clinical characteristics of the sample are presented in Table 2 . There were no significant demographic differences between case and control subjects, and no differences in several clinical variables that are known predictors of suicide (32) . Participants who developed suicidal ideation received a significantly higher maximum citalopram dose, were significantly less likely to go into remission (defined as a score £5 on the QIDS-C on the last recorded visit during citalopram treatment [Level 1]), and were more likely to move on to a secondary treatment phase (Level 2) after citalopram (15). Only 25% of those who had treatment-emergent suicidal ideation, compared with 42.9% of control subjects, achieved remission, although those with and without suicidal ideation did not differ significantly in initial symptom scores ( Table 1 ). Consistent with this poorer outcome, 62.5% of those with suicidal ideation went on to Level 2 to receive a change in treatment, compared with 44.9% of control subjects. There was no difference in the rates of exit from citalopram treatment.
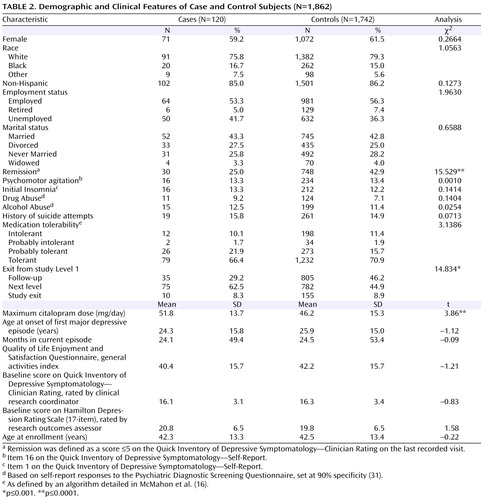
Consistent with previous reports (8) , treatment-emergent suicidal ideation developed relatively early in treatment: 21% of those who developed suicidal ideation did so by visit 2 (median=14 days after starting treatment), 69% by visit 3 (median=21 days), and 92% by visit 5 (median=28 days). By the end of Level 1, 48% of those who developed suicidal ideation had returned to their baseline score of 0, while 37% persisted, and 15% had a fluctuating course. None of the participants with treatment-emergent suicidal ideation are known to have attempted suicide.
Association Analysis
Figure 2 presents results of the allelic and genotypic association tests. Two markers produced significant evidence of association at the experimentwise p<0.05 level. Both markers were in Hardy-Weinberg equilibrium in this sample.
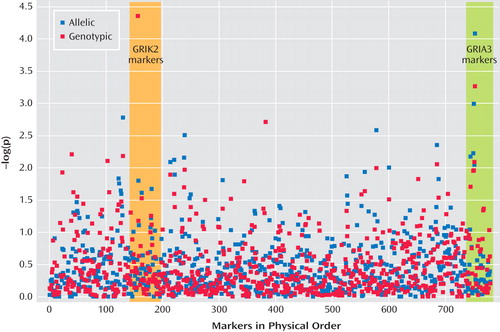
a Ordered by physical position along the autosomes (markers 1–719) and X-chromosome (markers 720–768) on Build 34 of the Draft Human Genome Sequence. The boxed areas encompass all the markers genotyped in GRIK2 and GRIA3.
A marker in the first intron of GRIK2 on chromosome 6 (marker rs2518224), which encodes the kainate-sensitive ionotropic glutamate receptor GluR6, was associated with treatment-emergent suicidal ideation in the genotypic test (CC genotype, nominal p=2.43×10 –5 , odds ratio=8.23; permutation p<0.003). This marker was not significantly associated in the allelic test.
A marker in the third intron of GRIA3 on chromosome X (marker rs4825476), which encodes the a-amino-5-hydroxy-3-methyl-4-isoxazole propionic acid-sensitive ionotropic glutamate receptor AMPA3, was associated with treatment-emergent suicidal ideation in the allelic test (G allele, nominal p=7.84×10 –5 , odds ratio=1.94; permutation p<0.01).
Regression Analysis
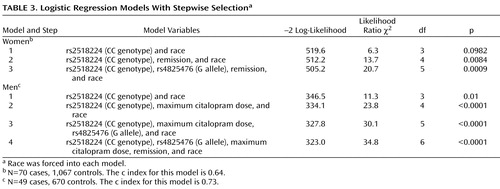
Having established experimentwise significant associations between two markers and treatment-emergent suicidal ideation in this sample, we next investigated the impact of nongenetic variables on the observed genetic association. By means of stepwise logistic regression, we tested the impact of race and the three clinical variables that had shown significant differences in those with and without treatment-emergent suicidal ideation in our initial analysis (see Table 2 ).
The best-fitting model was achieved with a combination of both markers, maximum citalopram dose, and remission by QIDS-C ( Table 3 ). Race was not a significant covariate in this model. Adjusted odds ratios were close to those in the unadjusted models. The Hosmer-Lemeshow test was nonsignificant for both genders, indicating a good model fit.
Population Structure
The association p values were uniformly distributed, with no excess of small values (Kolmogorov-Smirnov D=0.032, p=0.11). In the white subset, STRAT detected no evidence of mismatch between cases and controls (χ 2 =303.36, df=310, p=0.6).
Robustness to Varying Case Definition
The optimal case definition for treatment-emergent suicidal ideation is unknown. As a secondary analysis to assess the impact of varying case definition on the observed association findings, we examined treatment-emergent suicidal ideation as defined by the clinician-rated QIDS-C. The overall Pearson correlation of the QIDS-SR and QIDS-C scores on item 12 was low but highly significant (r=0.37, p=0.0001). The 144 cases of suicidal ideation defined by the QIDS-C alone showed association with markers in both GRIK2 and GRIA3 at the nominal p<0.01 level, although different single-nucleotide polymorphisms were involved. Participants who met the case definition for suicidal ideation on both the QIDS-SR and the QIDS-C (N=55) were significantly more likely to carry exactly the same marker alleles identified in our primary analysis than those who denied all suicidal thoughts on both instruments (χ 2 =15.42, df=2, p=0.0004).
Since our primary case definition included individuals who scored only 1 (“I feel that life is empty or wonder if it is worth living”) on the “thoughts of death or suicide” item, we compared allelic and genotypic frequencies of the GRIK2 and GRIA3 markers in those with treatment-emergent suicidal ideation who scored 1 and those who scored >1. There were no significant differences, which suggests that all patients who scored over 0 are similar with respect to allele frequencies at the GRIK2 and GRIA3 markers.
Combined Effect of Risk Alleles and Genotypes
Of the six combinations of high-risk alleles and genotypes tested, the highest odds ratio was observed in patients carrying both the high-risk allele of marker rs4825476 and the high-risk genotype of marker rs2518224 (odds ratio=14.98, 95% CI=3.7–60.674). Consistent with this, there was significant evidence of interaction between the two markers by the likelihood ratio test (χ 2 =12.3, df=1, p=0.0004). The combined impact of both markers on the risk of treatment-emergent suicidal ideation appears to be at least additive, but sample size limitations preclude any precise estimates of the mode of interaction.
Discussion
To our knowledge, this is the first study to detect a significant overall association between a genetic marker and treatment-emergent suicidal ideation. These data suggest that this uncommon but potentially dangerous adverse event may have a genetic component. Until functional alleles are demonstrated or replication is shown in an independent sample, these findings should be viewed as preliminary. However, they may shed light on the biological basis of suicidal ideation that emerges during antidepressant treatment and provide an initial step toward developing markers with clinically meaningful predictive value.
The validation of these findings through replication will be difficult, since treatment-emergent suicidal ideation is so uncommon (9) . Several thousand patients may need to be studied before a sufficiently large replication sample can be obtained. To our knowledge, the STAR*D sample is the only such sample available at this time. However, the association findings we report here show several hallmarks of causal associations (33) . The overall sample is large and is representative of major depression in the outpatient setting. The effect sizes are relatively large, and the statistical significance levels stand up to correction for the number of tests carried out. There is an apparent dose-response relationship between these markers and treatment-emergent suicidal ideation: more participants who carried both risk alleles reported suicidal ideation than those who carried only one allele. The implicated genes are biologically plausible candidates with closely related functions. Furthermore, association between these genes and treatment-emergent suicidal ideation persists across alternative case definitions.
This study has several limitations. Treatment-emergent suicidal ideation was defined on the basis of items in a depression rating scale, and neither the instrument nor the STAR*D study was designed to address this adverse event. Since there was no placebo group, we cannot determine what fraction of suicidal ideation in this study sample is directly attributable to antidepressant treatment. Moreover, the case definition we used is arbitrary. Given the lack of a widely accepted definition, we used one similar to that seen in the literature as well as that adopted by the FDA, which instituted the black box warning. Alternative case definitions are possible. We tested one alternative definition based on the QIDS-C, which yielded genetic association results similar to those we obtained with the definition based on QIDS-SR, and our results would remain significant at the p<0.05 level even if we doubled the permutation p values to account for a second possible phenotype definition. These limitations are characteristic of all existing large antidepressant treatment samples and highlight the need for large, placebo-controlled studies of treatment-emergent suicidal ideation in the future.
We observed that participants who developed suicidal ideation experienced a less favorable response to treatment overall, which could confound the interpretation of genetic association results. To address this limitation, we controlled for citalopram dose and remission rates in the secondary regression analysis. This analysis showed that the genetic markers we report were significant independent predictors of treatment-emergent suicidal ideation in this sample.
Another limitation of this particular study is that all participants were treated only with citalopram, so these findings may not be generalizable to other antidepressant medications. Citalopram is similar to other SSRIs and is one of the most widely prescribed antidepressants, so these findings have clinical relevance even if confined to citalopram and related compounds.
This study interrogated 68 genes. Although these genes were considered likely candidates for outcomes related to antidepressant treatment, there may be additional genes related to treatment-emergent suicidal ideation that were not studied here. For example, one previous study (34) found that markers in CREB1 were associated with treatment-emergent suicidal ideation in men. Moreover, the markers used did not cover the selected genes completely. Additional markers could detect additional association signals. Since markers were selected on the basis of low intermarker LD, haplotype tests would be expected to have poor power and thus were not performed. Still, this is the most comprehensive genetic study of treatment-emergent suicidal ideation conducted to date. More comprehensive studies will not likely emerge until after genome-wide association studies have been performed with this sample. As is true for most large studies, additional phenotypes have been and will continue to be tested over time in this sample. Our experiment-wide p value corrections, which are based on the hypotheses tested in this study, should be considered in this light.
Cryptic population stratification is always a risk in case-control genetic association studies. To address this, we showed that 1) there is no relationship between race and treatment-emergent suicidal ideation in this sample; 2) association p values were uniformly distributed within the largest (white) subset under the assumption of one population; and 3) there is no evidence of mismatch between participants with and without suicidal ideation within the white subset. Thus, these association results are not likely to be the result of population stratification.
While treatment-emergent suicidal ideation is an understandable cause for concern and has fueled widespread reassessment of antidepressant prescribing practices, it is not clear how it is related to suicidal behavior (9) . Evidence suggests that the risk of suicide attempt is higher in depressed persons before the prescription of antidepressants than afterward (35) . Only two participants attempted suicide while undergoing treatment in the STAR*D study, and one of them participated in DNA collection. Although this patient consistently denied suicidal ideation, he did carry the high-risk alleles for both GRIK2 and GRIA3.
The markers we have identified do not appear to be related to a general tendency toward suicide but rather to suicidal thoughts specifically emerging during antidepressant treatment. To our knowledge, neither of these genes has been previously associated with suicidal ideation. There was no difference in allele or genotype frequencies at either marker rs2518224 or marker rs4825476 in participants who had a history of suicide attempts or reported suicidal ideation at their initial visit before the start of treatment (data not shown).
Both markers implicated in this study lie within genes that encode ionotropic glutamate receptors. This is consistent with prior evidence that antidepressants affect glutamate signaling. Agonists to both ionotropic and metabotropic glutamate receptors may have an antidepressant-like effect (36) . Chronic antidepressant treatment increases membrane expression of AMPA receptors in rat hippocampus (37) in a time frame consistent with therapeutic effects. In addition, chronic treatment with SSRIs increases phosphorylation of the active sites of AMPA receptors in extracts of cortex, hippocampus, and striatum (38) . A recent study demonstrated specific and regionally discrete changes in the expression and editing of AMPA and kainate glutamate receptors, along with selective reduction of conductance for GluR3-containing receptors, after treatment with antidepressants (39) . That study also showed that prolonged exposure to antidepressants produced site-selective and area-specific effects on this particular posttranscriptional regulation.
In summary, we identified markers in two genes within the glutamate signaling pathway that may shed light on the biological basis of treatment-emergent suicidal ideation and have the potential to help identify patients at high risk of having suicidal ideation emerge during citalopram treatment. Such patients may benefit from closer monitoring, alternative treatments, or specialty care. Further work is needed to replicate these findings and uncover the functional variation that underlies the association signals we observed.
1. Teicher MH, Glod C, Cole JO: Emergence of intense suicidal preoccupation during fluoxetine treatment. Am J Psychiatry 1990; 147:207–210Google Scholar
2. Khan A, Khan S, Kolts R, Brown WA: Suicide rates in clinical trials of SSRIs, other antidepressants, and placebo: analysis of FDA reports. Am J Psychiatry 2003; 160:790–792Google Scholar
3. US Food and Drug Administration: Transcript of the US Food and Drug Administration Psychopharmacological Drugs Advisory Committee meeting, Sept 20, 1991. http://www.fda.gov/ohrms/dockets/ac/prozac/2443T1.pdfGoogle Scholar
4. Medicine and Healthcare Products Regulatory Agency: Safety review of antidepressants used by children (press release, reference 2003/0505). http://www.antidepressantsfacts.com/2003-10-12-05-05-MHRA-CSM-SSRIs.htmGoogle Scholar
5. US Food and Drug Administration: Antidepressant Use in Children, Adolescents and Adults, March 22, 2004. http://www.fda.gov/cder/drug/antidepressants/default.htmGoogle Scholar
6. Murray ML, Thompson M, Santosh PJ, Wong IC: Effects of the Committee on Safety of Medicines advice on antidepressant prescribing to children and adolescents in the UK. Drug Saf 2005; 28:1151–1157Google Scholar
7. US Food and Drug Administration: FDA public health advisory: suicidality in children and adolescents being treated with antidepressant medications, Oct 15, 2004. http://www.fda.gov/cder/drug/antidepressants/SSRIPHA200410.htmGoogle Scholar
8. Jick H, Kaye JA, Jick SS: Antidepressants and the risk of suicidal behaviors. JAMA 2004; 292:338–343Google Scholar
9. Licinio J, Wong ML: Depression, antidepressants, and suicidality: a critical appraisal. Nat Rev Drug Discov 2005; 4:165–171Google Scholar
10. Fava M, Rush AJ, Trivedi MH, Nierenberg AA, Thase ME, Sackeim HA, Quitkin FM, Wisniewski S, Lavori PW, Rosenbaum JF, Kupfer DJ: Background and rationale for the Sequenced Treatment Alternatives to Relieve Depression (STAR*D) study. Psychiatr Clin North Am 2003; 26:457–494Google Scholar
11. Rush AJ, Fava M, Wisniewski SR, Lavori PW, Trivedi MH, Sackeim HA, Thase ME, Nierenberg AA, Quitkin FM, Kashner TM, Kupfer DJ, Rosenbaum JF, Alpert J, Stewart JW, McGrath PJ, Biggs MM, Shores-Wilson K, Lebowitz BD, Ritz L, Niederehe G: Sequenced Treatment Alternatives to Relieve Depression (STAR*D): rationale and design. Control Clin Trials 2004; 25:119–142Google Scholar
12. Hamilton M: A rating scale for depression. J Neurol Neurosurg Psychiatry 1960; 23:56–62Google Scholar
13. Hamilton M: Development of a rating scale for primary depressive illness. Br J Soc Clin Psychol 1967; 6:278–296Google Scholar
14. American Psychiatric Association: Diagnostic and Statistical Manual of Mental Disorders, 4th ed. Washington, DC, American Psychiatric Association, 1994Google Scholar
15. Trivedi MH, Rush AJ, Wisniewski SR, Nierenberg AA, Warden D, Ritz L, Norquist G, Howland RH, Lebowitz B, McGrath PJ, Shores-Wilson K, Biggs MM, Balasubramani GK, Fava M, STAR*D Study Team: Evaluation of outcomes with citalopram for depression using measurement-based care in STAR*D: implications for clinical practice. Am J Psychiatry 2006; 163:28–40Google Scholar
16. McMahon FJ, Buervenich S, Charney D, Lipsky R, Rush AJ, Wilson AF, Sorant AJ, Papanicolaou GJ, Laje G, Fava M, Trivedi MH, Wisniewski SR, Manji H: Variation in the gene encoding the serotonin 2A receptor is associated with outcome of antidepressant treatment. Am J Hum Genet 2006; 78:804–814Google Scholar
17. Rush AJ, Carmody TJ, Reimitz P-E: The Inventory of Depressive Symptomatology (IDS): Clinician (IDS-C) and Self-Report (IDS-SR) ratings of depressive symptoms. Int J Methods Psychiatr Res 2000; 9:45–59Google Scholar
18. Rush AJ, Trivedi MH, Ibrahim HM, Carmody TJ, Arnow B, Klein DN, Markowitz JC, Ninan PT, Kornstein S, Manber R, Thase ME, Kocsis JH, Keller MB: The 16-item Quick Inventory of Depressive Symptomatology (QIDS), Clinician Rating (QIDS-C), and Self-Report (QIDS-SR): a psychometric evaluation in patients with chronic major depression. Biol Psychiatry 2003; 54:573–583Google Scholar
19. Trivedi MH, Rush AJ, Ibrahim HM, Carmody TJ, Biggs MM, Suppes T, Crismon ML, Shores-Wilson K, Toprac MG, Dennehy EB, Witte B, Kashner TM: The Inventory of Depressive Symptomatology, Clinician Rating (IDS-C) and Self-Report (IDS-SR), and the Quick Inventory of Depressive Symptomatology, Clinician Rating (QIDS-C) and Self-Report (QIDS-SR) in public sector patients with mood disorders: a psychometric evaluation. Psychol Med 2004; 34:73–82Google Scholar
20. Rush AJ, Trivedi MH, Carmody TJ, Ibrahim HM, Markowitz JC, Keitner GI, Kornstein SG, Arnow B, Klein DN, Manber R, Dunner DL, Gelenberg AJ, Kocsis JH, Nemeroff CB, Fawcett J, Thase ME, Russell JM, Jody DN, Borian FE, Keller MB: Self-reported depressive symptom measures: sensitivity to detecting change in a randomized, controlled trial of chronically depressed, nonpsychotic outpatients. Neuropsychopharmacology 2005; 30:405–416Google Scholar
21. Rush AJ, Bernstein IH, Trivedi MH, Carmody TJ, Wisniewski S, Mundt JC, Shores-Wilson K, Biggs MM, Woo A, Nierenberg AA, Fava M: An evaluation of the Quick Inventory of Depressive Symptomatology and the Hamilton Rating Scale for Depression: a Sequenced Treatment Alternatives to Relieve Depression trial report. Biol Psychiatry 2006; 59:493–501Google Scholar
22. Pritchard JK, Stephens M, Donnelly P: Inference of population structure using multilocus genotype data. Genetics 2000; 155:945–959Google Scholar
23. Pritchard JK, Stephens M, Rosenberg NA, Donnelly P: Association mapping in structured populations. Am J Hum Genet 2000; 67:170–181Google Scholar
24. Carlson CS, Eberle MA, Rieder MJ, Yi Q, Kruglyak L, Nickerson DA: Selecting a maximally informative set of single-nucleotide polymorphisms for association analyses using linkage disequilibrium. Am J Hum Genet 2004; 74:106–120Google Scholar
25. Gunderson KL, Kruglyak S, Graige MS, Garcia F, Kermani BG, Zhao C, Che D, Dickinson T, Wickham E, Bierle J, Doucet D, Milewski M, Yang R, Siegmund C, Haas J, Zhou L, Oliphant A, Fan JB, Barnard S, Chee MS: Decoding randomly ordered DNA arrays. Genome Res 2004; 14:870–877Google Scholar
26. Purcell S, Cherny SS, Sham PC: Genetic Power Calculator: design of linkage and association genetic mapping studies of complex traits. Bioinformatics 2003; 19:149–150Google Scholar
27. Sasieni PD: From genotypes to genes: doubling the sample size. Biometrics 1997; 53:1253–1261Google Scholar
28. Dudbridge F: Pedigree disequilibrium tests for multilocus haplotypes. Genet Epidemiol 2003; 25:115–221Google Scholar
29. Wigginton JE, Abecasis GR: PEDSTATS: descriptive statistics, graphics, and quality assessment for gene mapping data. Bioinformatics 2005; 21:3445–3447Google Scholar
30. Cordell HJ, Clayton DG: A unified stepwise regression procedure for evaluating the relative effects of polymorphisms within a gene using case/control or family data: application to HLA in type 1 diabetes. Am J Hum Genet 2002; 70:124–141Google Scholar
31. Rush AJ, Zimmerman M, Wisniewski SR, Fava M, Hollon SD, Warden D, Biggs MM, Shores-Wilson K, Shelton RC, Luther JF, Thomas B, Trivedi MH: Comorbid psychiatric disorders in depressed outpatients: demographic and clinical features. J Affect Disord 2005; 87:43–55Google Scholar
32. Hirschfeld RM, Russell JM: Assessment and treatment of suicidal patients. N Engl J Med 1997; 337:910–915Google Scholar
33. Hill AB: The environment and disease: association or causation? Proc R Soc Med 1965; 58:295–300Google Scholar
34. Perlis RH, Purcell S, Fava M, Fagerness J, Rush AJ, Trivedi MH, Smoller JW: Association between treatment-emergent suicidal ideation with citalopram and polymorphisms near cyclic adenosine monophosphate response element binding protein in the STAR*D study. Arch Gen Psychiatry 2007; 64:689–697Google Scholar
35. Simon GE, Savarino J, Operskalski B, Wang PS: Suicide risk during antidepressant treatment. Am J Psychiatry 2006; 163:41–47Google Scholar
36. Palucha A, Pilc A: The involvement of glutamate in the pathophysiology of depression. Drug News Perspect 2005; 18:262–268Google Scholar
37. Martinez-Turrillas R, Frechilla D, Del RJ: Chronic antidepressant treatment increases the membrane expression of AMPA receptors in rat hippocampus. Neuropharmacology 2002; 43:1230–1237Google Scholar
38. Svenningsson P, Tzavara ET, Witkin JM, Fienberg AA, Nomikos GG, Greengard P: Involvement of striatal and extrastriatal DARPP-32 in biochemical and behavioral effects of fluoxetine (Prozac). Proc Natl Acad Sci USA 2002; 99:3182–3187Google Scholar
39. Barbon A, Popoli M, La Via L, Moraschi S, Vallini I, Tardito D, Tiraboschi E, Musazzi L, Giambelli R, Gennarelli M, Racagni G, Barlati S: Regulation of editing and expression of glutamate alpha-amino-propionic-acid (AMPA)/kainate receptors by antidepressant drugs. Biol Psychiatry 2006; 59:713–720Google Scholar

