
Elevated Intrasynaptic Dopamine Release in Tourette’s Syndrome Measured by PET
Abstract
OBJECTIVE: Dopaminergic abnormalities in frontal-subcortical circuits have been hypothesized as the underlying pathophysiologic mechanism in Tourette’s syndrome. The objective of this study was to test the hypothesis that presynaptic dopamine release from the striatum is abnormal in adults with Tourette’s syndrome. METHOD: Seven adults with Tourette’s syndrome and five age-matched comparison subjects each received two positron emission tomography (PET) scans with high specific activity [11C]raclopride. The first scan followed an intravenous injection of saline; the second followed an intravenous injection of amphetamine. The relative dopamine release was estimated as the percentage difference in binding potential between the postsaline and postamphetamine scans. RESULTS: Binding potential determined after the initial [11C]raclopride scan did not significantly differ between Tourette’s syndrome and comparison subjects. After amphetamine challenge, the mean value of intrasynaptic dopamine in the putamen (as determined by true equilibrium bolus estimation) increased by 21% in the subjects with Tourette’s syndrome and did not change in the comparison subjects; the mean values increased by 16.9% and decreased by 1.8%, respectively, when measured by the constrained method. Dopamine release in the caudate region was not significantly different in the Tourette’s syndrome and comparison subjects. CONCLUSIONS: Greater putamen dopamine release was seen in adults with Tourette’s syndrome than in comparison subjects after a pharmacologic challenge with amphetamine. These results suggest that the underlying pathobiology in Tourette’s syndrome is a phasic dysfunction of dopamine transmission.
Tourette’s syndrome is an inherited neuropsychiatric disorder that appears in childhood and is characterized by the presence of chronic motor and vocal tics that have a waxing and waning course (1). Although the exact neuroanatomic localization of Tourette’s syndrome remains unknown, increasing evidence supports pathophysiologic involvement of frontal-subcortical circuits (2–5). On the basis of, in part, the distribution of classical neurotransmitters within these pathways, a variety of transmitters have been hypothesized to have a pathobiologic role in this syndrome.
The possibility of a dopaminergic abnormality in Tourette’s syndrome continues to receive strong consideration because of the therapeutic response to neuroleptics, preliminary data from postmortem studies, and a variety of nuclear imaging protocols (2, 6, 7). The present positron emission tomography (PET) study was designed to investigate dopamine release in adults with Tourette’s syndrome and a comparison group of healthy subjects.
Hypotheses invoking the role of dopamine in Tourette’s syndrome have suggested abnormalities of both post- and presynaptic function, such as supersensitive dopamine (D) receptors, dopamine hyperinnervation, and abnormal presynaptic terminal function. The possibility of abnormal postsynaptic receptors was initially suggested by findings of reduced basal and turnover levels of a dopamine metabolite, homovanillic acid, in cerebrospinal fluid that were restored to normal after the administration of haloperidol (6, 8, 9). Limited studies of D1 and D2 receptor binding in postmortem striatal tissue have shown differences between Tourette’s syndrome and comparison subjects, but none that reached significance (7). Investigations of D2 receptors by PET and single photon emission computed tomography (SPECT) have produced inconsistent findings in studies involving Tourette’s syndrome patients and comparison subjects (10–13). Two studies have supported the hypothesis that the dopamine receptor is involved in the neurobiology of Tourette’s syndrome. In a study of five sets of identical twins, greater [123I]iodobenzamide ([123I]-IBZM) binding observed in the head of the caudate nucleus was associated with greater tic severity (12). In our PET study with a spiperone derivative, [11C]3-N-methylspiperone, Bmax levels above the 95th percentile prediction limit (normal regressed against age) were observed in four of 20 adult subjects, and multiple linear regression analyses revealed a trend between the severity of vocal tics and Bmax values (13). In contrast to studies reporting D2 receptor changes, other studies in which [123I]-IBZM or [11C]raclopride was used did not show differences (10, 11).
Attempts to provide support for postulated dopamine hyperinnervation (i.e., greater number of dopamine terminals) by PET or SPECT binding have resulted in conflicting reports. For example, a small study that used [123I]β-CIT SPECT found that striatal dopamine transporter binding was higher in five adults with Tourette’s syndrome than in a group of comparison subjects (14). In a second study that used a similar technique, the mean striatal activity ratio was significantly higher in Tourette’s syndrome patients, and five of 12 Tourette’s syndrome subjects showed striatum/occipital cortex ratios more than two standard deviations above the normal mean (15). In contrast, other investigators who used SPECT or PET techniques in adult patients with Tourette’s syndrome reported no difference in dopamine transporter binding relative to that seen in comparison subjects (16, 17). Studies evaluating dorsal striatal dopaminergic innervation, by use of in vivo measures of vesicular monoamine transporter type 2 binding with the ligand (+)-alpha-[11C]dihydrotetrabenazine, showed no differences between Tourette’s syndrome subjects and age-compatible normal comparison subjects (18). These results suggest that there is no increase in striatal innervation but do not exclude an abnormality in the regulation of dopamine release or reuptake. Last, a third dopamine proposal implicates a presynaptic dopamine abnormality involving dopa decarboxylase activity. In a PET study, 11 adolescents with Tourette’s syndrome accumulated [18F]fluorodopa at a level 25% higher in the left caudate nucleus and 53% higher in the right midbrain relative to levels seen in comparison subjects (19). The authors suggested that upregulation of dopa decarboxylase activity could explain these alterations and that the process reflects deficits in a variety of functional elements of the dopamine system.
In order to evaluate dopamine release in Tourette’s syndrome, we intravenously administered a central stimulant (amphetamine) that enhances dopamine release and blocks its reuptake (20). Analogous studies with such a provocative stimulant challenge have been performed in rodents (21), baboons (22), normal humans (23), and in patients with schizophrenia (24, 25) and cocaine dependence (26). In the current study, each subject underwent two [11C]raclopride PET scans: one after intravenous administration of saline and the second after amphetamine was intravenously administered to induce dopamine release. Studies that have used a stimulant challenge followed by either PET or SPECT binding have demonstrated greater dopamine release in individuals with schizophrenia (24, 25) and decreased release in those with cocaine dependence (27). Application of the amphetamine stimulation technique to adults with Tourette’s syndrome suggests that tics may be associated with higher intrasynaptic levels of dopamine.
Method
Subjects
Seven adult male subjects with Tourette’s syndrome (mean age=37 years, range=19 to 50) were voluntary participants. This study was approved by the Johns Hopkins University School of Medicine’s Joint Committee on Clinical Investigation, and written informed consent was obtained after complete description of the study to the subjects. All fulfilled the diagnostic criteria for Tourette’s syndrome as defined by the Tourette Syndrome Classification Study Group (28). The mean age of tic onset was 6 years (range=3–12). Four (57%) reported a family history of tics. Lifetime global tic severity was determined for each subject on the basis of history, clinical observation, and two tic severity scales (the Yale Global Tic Severity Scale [29] and the Hopkins Motor and Vocal Tic Severity Scale [30]). The seven subjects had global lifetime rankings of mild/moderate (N=2), moderate (N=2), moderate/severe (N=2), and severe (N=1). The mean baseline total score on the Yale Global Tic Severity Scale obtained on the day of the PET scan was 38 (SD=9, range=20–45). No subject was receiving medication at the time of the study. No patient had received tic-suppressing medication within the 6 months before the PET study. Two subjects had never received tic-suppressing medication, whereas three had received prior treatment with neuroleptics. All Tourette’s syndrome subjects had nonfocal neurological examination results and normal levels of cognitive functioning as determined by clinical assessment. Six patients reported a history consistent with attention deficit hyperactivity disorder (ADHD), but no additional attempt was made to confirm this diagnosis. Obsessive-compulsive disorder (OCD), evaluated by a structured interview, was diagnosed in two (29%) of the seven subjects.
The tic-free comparison group consisted of five age-matched adults (three men and two women; mean age=40 years, range=20–53) recruited from hospital personnel and the local community by newspaper advertisements. All subjects provided a medical history and underwent a physical examination; one subject reported a history of alcohol dependency but denied current use.
Imaging Protocols
Each subject underwent a magnetic resonance imaging (MRI) scan before the PET scan to ensure accurate measurement of regional radiotracer activity. A spoiled gradient recall volumetric acquisition with a T1-weighted sagittal scout image was obtained for identification of the anterior commissure-posterior commissure line. A form-fitted thermal plastic facial mask with laser markings was used to guarantee similar head placement during both the MRI and PET scans.
Each subject underwent two PET scans with high specific activity [11C]raclopride. The first scan followed an intravenous injection of saline; the second scan followed an intravenous injection of amphetamine. This fixed sequence of injections mimics scanning protocols used in studies of individuals with schizophrenia and cocaine abuse (24, 25, 27). Both studies were performed on the same day with a time interval of about 4.5 hours between scans. Equal volumes of either saline or amphetamine (0.3 mg/kg) were administered intravenously over 90 seconds, 5 minutes before the raclopride injection. Approximately 20 mCi of [11C]raclopride (range=17.99 to 21.82; mass <10 μg) was administered by intravenous bolus injection over 20 seconds. No significant differences in raclopride mass per body weight were seen between Tourette’s syndrome and healthy comparison subjects.
PET images were acquired on a GE 4096 Plus PET scanner. Each PET scan consisted of 50 sequences, ranging from 15 seconds to 6 minutes in duration, and 15 contiguous 6-mm thick transaxial brain slices over a 90-minute total acquisition period. A resolution of approximately 6 mm full width at half maximum was attained by data reconstruction with a Hanning filter in a VAX computer. All scans had attenuation correction by use of the built-in rotating pin source (10 mCi of Ge68) of the 4096 Plus PET scanner. MRI-PET registration was performed by using a UNIX workstation and the computer program Register (31). Regions of interest were drawn on multiple planes and then averaged.
Blood samples for plasma radioactive time curves were obtained from a radial artery catheter with collection intervals ranging from 3 seconds to 15 minutes over the 90-minute scanning. Plasma was separated by centrifugation, and aliquots were assayed in a gamma counter. The radioactive time curve was corrected for the presence of radiolabeled metabolites. [11C]Raclopride metabolites were assayed on blood samples at 5, 12, 20, 30, 45, 60, and 75 minutes after the injection by high-performance liquid chromatography on a reverse-phase high-performance liquid chromatography column (32).
Data Analysis: Parametric Analysis of [11C]Raclopride Distribution
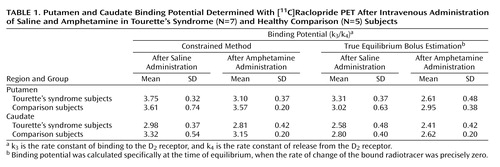
Kinetic modeling analysis of the rate constants k1, k2, k3, and k4, were obtained by using a four-parameter model as previously described (13): k1 is the rate of clearance of ligand from the circulation; k2 is the fractional clearance of ligand from the region of interest to the plasma; k3 is the rate constant of binding to the D2 receptor; and k4 is the rate constant of release from the D2 receptor. The k1/k2 ratio was constrained to be equal in the cerebellum and putamen, and the k3/k4 ratio was examined in the putamen and caudate. The relative dopamine release was estimated as the percentage difference in binding potential (Bmax/KD), calculated as the k3/k4 ratio, between postsaline and postamphetamine scans. One Tourette’s syndrome subject was evaluated by using a two-compartment model with the cerebellum as input, since only arterialized venous blood was available for determination of the plasma radioactive time curve (Figure 1).
Results
The mean binding potentials following the initial (postsaline) and repeat (postamphetamine) [11C]raclopride scans in Tourette’s syndrome and healthy comparison subjects are presented in Table 1. Comparisons of the mean postsaline results in Tourette’s syndrome and comparison subjects, measured by the constrained method and true equilibrium bolus estimation (20, 33), showed no significant differences.
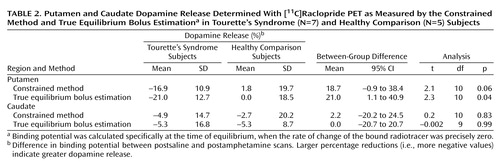
Dopamine release induced by amphetamine was calculated by determining the percentage difference in binding potential between the postsaline and postamphetamine scans; larger percentage reductions indicate greater dopamine release (Figure 2). Relative dopamine release in the putamen as measured by true equilibrium bolus estimation for the Tourette’s syndrome and healthy comparison subjects was significantly different (Table 2), and this difference approached significance when the constrained method was used. Changes in dopamine release did not correlate with patient age, global tic severity ranking, baseline Yale Global Tic Severity Scale, or presence of OCD. Relative dopamine release in the caudate, as measured by either the constrained method or true equilibrium bolus estimation, did not significantly differ between the Tourette’s syndrome and healthy comparison subjects.
Discussion
Dopamine has an important influence on frontal-subcortical neurotransmission, either through presynaptic effects on corticostriatal afferents or postsynaptic effects on striatal neurons. In Tourette’s syndrome, dopamine hypotheses have been proposed that suggest either an excess of dopamine or an increased sensitivity to the neurotransmitter. Although several SPECT and PET studies have suggested that some individuals may have supersensitive D2 receptors (12, 13), concerns about these investigations include the effect of prior pharmacotherapy and the lack of compelling evidence that this is the primary abnormality in the majority of subjects. In the present investigation, similar to a prior PET study with [11C]raclopride (11) and a SPECT study with [123I]-IBZM (10), mean binding potential following the saline infusion did not significantly differ between Tourette’s syndrome and comparison subjects. These results could be interpreted as reflecting normal D2 receptor levels; however, several investigators have claimed that benzamide radioligands have greater susceptibility to endogenous dopamine that may, in turn, mask Bmax differences (34, 35). A second hypothesis, which proposes dopaminergic hyperinnervation, is negated in part by findings of normal presynaptic striatal dopamine terminals as determined by quantifying regional densities of dopamine transporter and vesicular monoamine transporter type 2 (17, 18). Nevertheless, although there may be normal numbers of dopamine terminals, a functional alteration of dopamine reuptake or dopamine release remains possible.
Volkow and colleagues (23, 27) pioneered the use of a methylphenidate plus [11C]raclopride PET binding method to measure intrasynaptic dopamine content and subsequently showed levels to be diminished in cocaine users. Laruelle et al. (24) substituted intravenous amphetamine for methylphenidate and, using [123I]-IBZM SPECT, reported significantly greater dopamine release in adult schizophrenic patients than in normal comparison subjects. The fixed scan order (saline first, then amphetamine) is the standard protocol for all such bolus studies and is not considered a potential confound (23, 36, 37). In fact, the initial use of amphetamine could result in a possible carryover effect and a necessary prolonged delay before the saline study. A smaller magnitude of change in our comparison subjects, relative to that reported in other protocols (24, 25, 38), is believed to be due to our use of a bolus injection of amphetamine that was not followed by a constant infusion.
The present study suggests that after a stimulant challenge, there is a greater release of dopamine in the putamen of adults with Tourette’s syndrome relative to that seen in comparison subjects. Although the total number of subjects in our study is limited, the comparison group had dopamine release measures in the putamen consistent with prior reports that used an amphetamine [123I]-IBZM SPECT paradigm (24). In the putamen of Tourette’s syndrome subjects, values of intrasynaptic dopamine as measured by true equilibrium bolus estimation increased by 21% after amphetamine challenge, which was significantly larger than that seen in the comparison group. The difference in relative dopamine release in the putamen between Tourette’s syndrome and comparison subjects approached significance when the less sensitive constrained method of analysis was used. Although the change in dopamine release appears unrelated to age, tic severity, and OCD, whether it is associated with other symptoms, such as ADHD, remains unclear. Dopamine transporter density, measured by SPECT, has been shown to be greater in adults with ADHD than in healthy comparison subjects (39). Although global tic severity ranking did not correlate with the degree of dopamine release, potential explanations include the small number of study subjects and characteristics relating to tics, i.e., their natural variability, possible improvement over time, and exacerbation by external factors such as stress, anxiety, fatigue, or the presence of infection (40–42).
Intrasynaptic release of dopamine in the caudate of Tourette’s syndrome subjects was not significantly different from that of the comparison subjects. It is possible that differences in the caudate were partially obscured by methodological issues—a greater effect of partial volume due to a smaller caudate area and the finite resolution of the PET scanner. It is also possible that, consistent with volumetric MRI studies that have shown changes in symmetry or size of the putamen but little alteration of the caudate (43, 44), the putamen is the primary site of pathology in Tourette’s syndrome.
Although similar differences in putamen dopamine release were recorded when either true equilibrium bolus estimation or the constrained method of measurement was used, differences as measured by true equilibrium bolus estimation had greater significance. This is consistent with the belief that true equilibrium bolus estimation is a more optimal representation of binding potential and, in turn, the measurement of intrasynaptic dopamine release. In this relatively new method, binding potential and Bmax, when used with a low specific activity injection, is calculated specifically at the time of equilibrium when the rate of change of the bound radiotracer is precisely zero following a bolus injection (13, 33). Of the various methods for obtaining binding potential after a bolus injection, true equilibrium bolus estimation is considered the most appropriate.
Explanations for the higher intrasynaptic dopamine levels in the putamen of Tourette’s syndrome subjects following amphetamine stimulation are speculative. Among several possibilities are an increase in release of dopamine from the presynaptic terminal secondary to a localized defect in the release mechanism; a lack of presynaptic inhibition; an increase in firing of presynaptic neurons; or a functional defect in dopamine reuptake from the synaptic cleft. The modulation of dopamine release involves a complex interaction of reciprocal autoreceptor and heteroreceptor control (45, 46). For example, the release of dopamine is inhibited by activation of α2 adrenergic receptors (47, 48) and facilitated by serotonin-1A receptors (45). Furthermore, although levels of dopamine transporter binding may be affected in only some patients with Tourette’s syndrome (15), a functional abnormality of dopamine reuptake from the synaptic cleft could elevate dopamine levels in synaptic vesicles, and, in turn, increase dopamine release.
An alternative unifying hypothesis could lie in the tonic-phasic model of dopamine release (49). Based on electrophysiological and neurochemical data, two types of dopamine kinetics exist in the extracellular space. Tonic dopamine, which exists extracellularly in low concentration, determines the long-term or homeostatic mechanism. This “basal” level of dopamine is defined primarily as an extrasynaptic measure and calculated by use of microdialysis and electrophysiologic measurements. Dopamine autoreceptors (D2 and D3 subtypes) are proposed as regulators of tonic dopamine control (46). Phasic dopamine is the spike-dependent dopamine released primarily into the synapse. It can escape the synaptic cleft with sufficient stimulation or when an uptake blocker is given in high concentrations. Intrasynaptic dopamine release induced by the use of stimulant challenges, such as with amphetamine, has been proposed as a surrogate measurement for phasic dopamine. The tonic-phasic model has been used to explain an increase in dopamine release observed in patients with schizophrenia (24, 25, 49, 50).
Clinical and imaging studies, including our dopamine release data, are consistent with the possibility that the underlying pathobiology in Tourette’s syndrome is an abnormal regulation of the phasic dopamine response resulting in a hyperresponsive spike-dependent dopaminergic system. Two basic mechanisms are proposed for alteration of the phasic dopamine. The first is a lower sensitivity of phasic dopamine to tonic stimulation. This is unlikely, however, in view of the apparent marked responsiveness to a systemic dopamine agonist, as shown by the increase in dopamine release following our amphetamine challenge. A second possibility is a decrease in tonic dopamine levels. We favor this latter concept on the basis of lower levels of CSF HVA (6, 8, 9) and evidence for elevated D2 receptors in some subgroups of Tourette’s syndrome patients (13). If this is indeed the case, how does the decrease in tonic dopamine occur? Diminished tonic dopamine levels could be secondary to a decrease in phasic overflow from the synaptic cleft to the extracellular space, but this is not likely because it conflicts with the observation of an increase in phasic dopamine release. A decrease in tonic dopamine could be secondary to a diminished cortical afferent input, i.e., tonic dopamine release is regulated by cortical glutamatergic afferents (49). To our knowledge, however, postmortem and neuroimaging studies have not identified reduced cortical gray matter volume or abnormal efferent output. Anderson et al. (51) evaluated postmortem 13 brain regions from four Tourette’s syndrome patients and reported lower glutamate levels in the globus pallidus and substantia nigra pars reticulata but not in the putamen. Last, a decrease in tonic dopamine levels could be secondary to an increase in activity of the dopamine transporter, since the reuptake transporter determines the concentration of extrasynaptic dopamine. Hence, we propose that the essential underlying mechanism in Tourette’s syndrome could be an overactive dopamine transporter system. This situation would create reduced levels of extracellular dopamine, higher concentrations of dopamine in the axon terminal, an increase in stimulus-dependent dopamine release, autoreceptor supersensitivity at the presynaptic site, and an increase in sensitivity to low-dose neuroleptics. Several clinical findings in Tourette’s syndrome patients support the overactive dopamine transporter hypothesis. For example, the exacerbation of tics by stimulant medications (52, 53) could be secondary to greater dopamine release from the axon terminal. Environmental stimuli, such as stress, anxiety, and medications, well known to exacerbate tics, have been shown to increase phasic bursts of dopamine. Last, tic suppression with very low doses of neuroleptics (54) may occur because there is less tonic dopamine available for the neuroleptic to block.
Future PET studies involving larger numbers of Tourette’s syndrome subjects and using techniques in which measurements of D2 receptors, dopamine transporter, and dopamine release are available for each subject should provide further clarification of the dopaminergic system in adults with Tourette’s syndrome. Confirmation of a release abnormality in larger numbers of patients with Tourette’s syndrome could, in turn, lead to the development of new tic-suppressing pharmacotherapies.
 |
 |
Received Aug. 21, 2001; revision received Feb. 27, 2002; accepted March 13, 2002. From the Departments of Neurology, Pediatrics, Radiology, and Psychiatry, Johns Hopkins University School of Medicine; and the University of Pittsburgh Departments of Neuroscience and Psychiatry, Pittsburgh. Address reprint requests to Dr. Wong, Department of Radiology, Johns Hopkins Medical Institutions, JHOC 3245, 601 North Caroline St., Baltimore, MD 21287-0807; [email protected] (e-mail). Supported in part by a grant from the Tourette Syndrome Association and NIH grants NS-38927, MH-42821, DA-09482, and HD-24061. The authors thank the members of the PET Center staff, including R.F. Dannels, H. Ravert, D. Clough, J. Musachio, and W. Matthews.

Figure 1. Typical Putamen and Cerebellum Plasma Radioactivity During [11C]Raclopride PET After Intravenous Administration of Saline and Amphetamine in Tourette’s Syndrome (N=7) and Healthy Comparison (N=5) Subjectsa
aPlasma radioactivity time curves were determined by using a two-compartment model with the cerebellum as input, since only arterialized venous blood was available for evaluation. The time curves above are from a 45-year-old male subject with Tourette’s syndrome.

Figure 2. Putamen Dopamine Release Determined With [11C]Raclopride PET as Measured by True Equilibrium Bolus Estimationa in Healthy Comparison and Tourette’s Syndrome Subjects
aBinding potential was calculated specifically at the time of equilibrium, when the rate of change of the bound radiotracer was precisely zero.
bDifference in binding potential between postsaline and postamphetamine scans. Larger percentage reductions (i.e., more negative values) indicate greater dopamine release.
1. Singer HS, Walkup JT: Tourette syndrome and other tic disorders: diagnosis, pathophysiology, and treatment. Medicine 1991; 70:15-32Crossref, Medline, Google Scholar
2. Singer HS: Neurobiology of Tourette syndrome. Neurol Clin 1997; 15:357-379Crossref, Medline, Google Scholar
3. Singer HS: Current issues in Tourette syndrome. Mov Disord 2000; 15:1051-1063Crossref, Medline, Google Scholar
4. Peterson BS, Skudlarski P, Anderson AW, Zhang H, Gatenby JC, Lacadie CM, Leckman JF, Gore JC: A functional magnetic resonance imaging study of tic suppression in Tourette syndrome. Arch Gen Psychiatry 1998; 55:326-333Crossref, Medline, Google Scholar
5. Stern E, Silbersweig DA, Chee KY, Holmes A, Robertson MM, Trimble M, Frith CD, Frackowiak RS, Dolan RJ: A functional neuroanatomy of tics in Tourette syndrome. Arch Gen Psychiatry 2000; 57:741-748Crossref, Medline, Google Scholar
6. Singer HS, Butler IJ, Tune LE, Seifert WE, Coyle JT: Dopaminergic dysfunction in Tourette syndrome. Ann Neurol 1982; 12:361-366Crossref, Medline, Google Scholar
7. Singer HS, Hahn I-H, Moran TH: Abnormal dopamine uptake sites in postmortem striatum from patients with Tourette’s syndrome. Ann Neurol 1991; 30:558-562Crossref, Medline, Google Scholar
8. Butler IJ, Koslow SH, Seifert WE Jr, Caprioli RM, Singer HS: Biogenic amine metabolism in Tourette syndrome. Ann Neurol 1979; 6:37-39Crossref, Medline, Google Scholar
9. Cohen DJ, Shaywitz BA, Caparulo BK, Young JG, Bowers MB Jr: Chronic, multiple tics of Gilles de la Tourette’s disease: CSF acid monoamine metabolites after probenecid administration. Arch Gen Psychiatry 1978; 35:245-250Crossref, Medline, Google Scholar
10. George MS, Robertson MM, Costa DC, Ell PJ, Trimble MR, Pilowsky L, Verhoeff NP: Dopamine receptor availability in Tourette’s syndrome. Psychiatry Res 1994; 55:193-203Crossref, Medline, Google Scholar
11. Turjanski N, Sawle GV, Playford ED, Weeks R, Lammerstma AA, Lees AJ, Brooks DJ: PET studies of the presynaptic and postsynaptic dopaminergic system in Tourette syndrome. J Neurol Neurosurg Psychiatry 1994; 57:688-692Crossref, Medline, Google Scholar
12. Wolf SS, Jones DW, Knable MB, Gorey JG, Lee KS, Hyde TM, Coppola R, Weinberger DR: Tourette syndrome: prediction of phenotypic variation in monozygotic twins by caudate nucleus D2 receptor binding. Science 1996; 273:1225-1227Crossref, Medline, Google Scholar
13. Wong DF, Singer HS, Brandt J, Shaya E, Chen C, Brown J, Kimball AW, Gjedde A, Dannals RF, Ravert HT, Wilson PD, Wagner HN Jr: D2-like dopamine receptor density in Tourette syndrome measured by PET. J Nucl Med 1997; 38:1243-1247Medline, Google Scholar
14. Malison RT, McDougle CJ, van Dyck CH, Scahill L, Baldwin RM, Seibyl JP, Price LH, Leckman JF, Innis RB: [123I]β-CIT SPECT imaging of striatal dopamine transporter binding in Tourette’s disorder. Am J Psychiatry 1995; 152:1359-1361Link, Google Scholar
15. Muller-Vahl KR, Berding G, Brucke T, Kolbe H, Meyer GJ, Hundeshagen H, Dengler R, Knapp WH, Emrich HM: Dopamine transporter binding in Gilles de la Tourette syndrome. J Neurol 2000; 247:514-520Crossref, Medline, Google Scholar
16. Heinz A, Knable MB, Wolf SS, Jones DW, Gorey JG, Hyde TM, Weinberger DR: Tourette’s syndrome: [I-123]beta-CIT SPECT correlates of vocal tic severity. Neurology 1998; 51:1069-1074Crossref, Medline, Google Scholar
17. Wong DF, Ricaurte G, Grunder G, Rothman R, Naidu S, Singer H, Harris J, Yokoi F, Villemagne V, Szymanski S, Gjedde A, Kuhar M: Dopamine transporter changes in neuropsychiatric disorders. Adv Pharmacol 1998; 42:219-223Crossref, Medline, Google Scholar
18. Meyer P, Bohnen NI, Minoshima S, Koeppe RA, Wernette K, Kilbourn MR, Kuhl DE, Frey KA, Albin RL: Striatal presynaptic monoaminergic vesicles are not increased in Tourette’s syndrome. Neurology 1999; 53:371-374Crossref, Medline, Google Scholar
19. Ernst M, Zametkin AJ, Jons PH, Matochik JA, Pascualvaca D, Cohen RM: High presynaptic dopaminergic activity in children with Tourette’s disorder. J Am Acad Child Adolesc Psychiatry 1999; 38:86-94Crossref, Medline, Google Scholar
20. Wong DF, Sølling T, Yokoi F, Gjedde A: Quantification of extracellular dopamine release in schizophrenia and cocaine use by means of TREMBLE, in Quantitative Functional Brain Imaging With Positron Emission Tomography. Edited by Carson R, Daube-Witherspoon M, Herscovitch P. New York, Academic Press, 1998, pp 463-468Google Scholar
21. Young LT, Wong DF, Goldman S, Minkin E, Chen C, Matsumura K, Scheffel U, Wagner HN Jr: Effects of endogenous dopamine on kinetics of [3H]N-methylspiperone and [3H]raclopride binding in rat brain. Synapse 1991; 9:188-194Crossref, Medline, Google Scholar
22. Dewey SL, Smith GS, Logan J, Brodie JD, Fowler JS, Wolf AP: Striatal binding of the PET ligand [11C]raclopride is altered by drugs that modify synaptic dopamine levels. Synapse 1993; 13:350-356Crossref, Medline, Google Scholar
23. Volkow ND, Wang G-J, Fowler JS, Logan J, Schyler D, Hitzemann R, Lieberman J, Angrist B, Pappas N, MacGregor R: Imaging endogenous dopamine competition with [11C]raclopride in the human brain. Synapse 1994; 16:255-262Crossref, Medline, Google Scholar
24. Laruelle M, Abi-Dargham A, van Dyck CH, Gil R, D’Souza CD, Erdos J, McCance E, Rosenblatt W, Fingado C, Zoghbi SS, Baldwin RM, Seibyl JP, Krystal JH, Charney DS, Innis RB: Single photon emission computerized tomography imaging of amphetamine-induced dopamine release in drug-free schizophrenic subjects. Proc Natl Acad Sci USA 1996; 93:9235-9240Crossref, Medline, Google Scholar
25. Breier A, Su T-P, Saunders R, Carson RE, Kolachana BS, de Bartolomeis A, Weinberger DR, Weisenfeld N, Malhotra AK, Eckelman WC, Pickar D: Schizophrenia is associated with elevated amphetamine-induced synaptic dopamine concentrations: evidence from a novel positron emission tomography method. Proc Natl Acad Sci USA 1997; 94:2569-2574Crossref, Medline, Google Scholar
26. Schlaepfer TE, Pearlson GD, Wong DF, Marenco S, Dannals RF: PET study of competition between intravenous cocaine and [11C]raclopride at dopamine receptors in human subjects. Am J Psychiatry 1997; 154:1209-1213Link, Google Scholar
27. Volkow ND, Gatley SJ, Fowler JS, Chen R, Logan J, Dewey SL, Ding YS, Pappas N, King P, MacGregor RR, Kuhar MJ, Carroll FI, Wolf AP: Long-lasting inhibition of in vivo cocaine binding to dopamine transporters by 3 beta-(4-iodophenyl)tropane-2-carboxylic acid methyl ester: RTI-55 or beta CIT. Synapse 1995; 19:206-211Crossref, Medline, Google Scholar
28. The Tourette Syndrome Classification Study Group: Definitions and classification of tic disorders. Arch Neurol 1993; 50:1013-1016Crossref, Medline, Google Scholar
29. Leckman JF, Riddle MA, Hardin MT, Ort SI, Swartz KL, Stevenson J, Cohen J: The Yale Global Tic Severity Scale: initial testing of a clinician-rated scale of tic severity. J Am Acad Child Adolesc Psychiatry 1989; 28:566-573Crossref, Medline, Google Scholar
30. Walkup JT, Rosenberg LA, Brown J, Singer HS: The validity of instruments measuring tic severity in Tourette syndrome. J Am Acad Child Adolesc Psychiatry 1992; 31:472-477Crossref, Medline, Google Scholar
31. Evans AC, Marrett S, Torrescorzo J, Ku S, Collins L: MRI-PET correlation in three dimensions using a volume-of-interest (VOI) atlas. J Cereb Blood Flow Metab 1991; 11:A69-A78Google Scholar
32. Hilton J, Yokoi F, Dannals RF, Ravert HT, Szabo Z, Wong DF: Column-switching HPLC for the analysis of plasma in PET imaging studies. Nucl Med Biol 2000; 27:627-630Crossref, Medline, Google Scholar
33. Sølling T, Brust P, Cunningham V, Wong DF, Gjedde A: True equilibrium bolus estimation (TREMBLE) confirms rapid transient equilibrium (abstract). Neuroimage 1998; 5:A29Google Scholar
34. Wong DF, Young D, Wilson PD, Meltzer CC, Gjedde A: Quantification of neuroreceptors in the living human brain, III: D2-like dopamine receptors: theory, validation and changes during normal aging. J Cereb Blood Flow Metab 1997; 17:316-330Crossref, Medline, Google Scholar
35. Laruelle M: Imaging synaptic neurotransmission with in-vivo binding competition techniques: a critical review. J Cereb Blood Flow Metab 2000; 20:423-451Crossref, Medline, Google Scholar
36. Smith GS, Dewey SL, Brodie JD, Logan J, Vitkun SA, Simkowitz P, Schloesser R, Alexoff DA, Hurley A, Cooper T, Volkow ND: Serotonergic modulation of dopamine measured with [11C]raclopride and PET in normal human subjects. Am J Psychiatry 1997; 154:490-496Link, Google Scholar
37. Drevets WC, Gautier C, Price JC, Kupfer DJ, Kinahan PE, Grace AA, Price JL, Mathis CA: Amphetamine-induced dopamine release in human ventral striatum correlates with euphoria. Biol Psychiatry 2001; 49:81-96Crossref, Medline, Google Scholar
38. Abi-Dargham A, Gil R, Krystal J, Baldwin RM, Seibyl JP, Bowers M, van Dyck CH, Charney DS, Innis RB, Laruelle M: Increased striatal dopamine transmission in schizophrenia: confirmation in a second cohort. Am J Psychiatry 1998; 155:761-767Link, Google Scholar
39. Dougherty DD, Bonab AA, Spencer TJ, Rauch SL, Madras BK, Fischman AJ: Dopamine transporter density in patients with attention deficit hyperactivity disorder. Lancet 1999; 354:2132-2133Crossref, Medline, Google Scholar
40. Leckman JF, Zhang H, Vitale A, Lahnin F, Lynch K, Bondi C, Kim YS, Peterson BS: Course of tic severity in Tourette syndrome: the first two decades. Pediatrics 1998; 102:14-19Crossref, Medline, Google Scholar
41. Goetz CG, Tanner CM, Stebbins GT, Leipsig G, Carr WC: Adult tics in Gilles de la Tourette’s syndrome: descriptions and risk factors. Neurology 1992; 42:784-788Crossref, Medline, Google Scholar
42. Singer HS, Giuliano JD, Zimmerman AM, Walkup JT: Infection: a stimulus for tic disorders. Pediatr Neurol 2000; 22:380-383Crossref, Medline, Google Scholar
43. Singer HS, Reiss AL, Brown JE, Aylward EH, Shih B, Chee E, Harris EL, Reader MJ, Chase GA, Bryan RN: Volumetric MRI changes in basal ganglia of children with Tourette’s syndrome. Neurology 1993; 43:950-956Crossref, Medline, Google Scholar
44. Peterson B, Riddle MA, Cohen DJ, Katz LD, Smith JC, Hardin MT, Leckman JF: Reduced basal ganglia volumes in Tourette’s syndrome using three-dimensional reconstruction techniques from magnetic resonance images. Neurology 1993; 43:941-949Crossref, Medline, Google Scholar
45. Gobert A, Rivet J-M, Audinot V, Newman-Tancredi A, Cistarelli L, Millan MJ: Simultaneous quantification of serotonin, dopamine and noradrenaline levels in single frontal cortex dialysates of freely-moving rats reveals a complex pattern of reciprocal auto- and heteroreceptor-mediated control of release. Neuroscience 1998; 84:413-429Crossref, Medline, Google Scholar
46. Gobert A, Rivet J-M, Audinot V, Cistarelli L, Spedding M, Vian J, Peglion JL, Millan MJ: Functional correlates of dopamine D3 receptor activation in rat in vivo and their modulation by the selective antagonist, (+)-S 14297 II: both D2 and “silent” D3 autoreceptors control synthesis and release in mesolimbic, mesocortical and nigrostriatal pathways. J Pharmacol Exp Ther 1995; 275:899-913Medline, Google Scholar
47. Yavich L, Lappalainen R, Sirvio J, Haapalinna A, MacDonald E: α2-Adrenergic control of dopamine overflow and metabolism in mouse striatum. Eur J Pharmacol 1997; 339:113-119Crossref, Medline, Google Scholar
48. Trendelenburg A-U, Starke K, Limberger N: Presynaptic α2A-adrenoceptors inhibit the release of endogenous dopamine in rabbit caudate nucleus slices. Naunyn Schmeidebergs Arch Pharmacol 1994; 350:473-481Crossref, Medline, Google Scholar
49. Grace AA: Cortical regulation of subcortical dopamine systems and its possible relevance to schizophrenia. J Neural Transm Gen Sect 1993; 91:111-134Crossref, Medline, Google Scholar
50. Wong DF, Giedde A, Reith J, Grunder G, Szymanski S, Yokoi F, Hong C, Nestadt G, Neufeld G, Pearlson G, Tune L, Andrist B: Imaging intrasynaptic, postsynaptic and presynaptic dopamine dysfunction in psychosis (abstract). Soc Neurosci 1997; 23:1405Google Scholar
51. Anderson GM, Pollak ES, Chatterjee D, Leckman JF, Riddle MA, Cohen DJ: Brain monoamines and amino acids in Gilles de la Tourette’s syndrome: a preliminary study of subcortical regions. Arch Gen Psychiatry 1992; 49:584-586Crossref, Medline, Google Scholar
52. Erenberg G, Cruse RP, Rothner AD: Gilles de la Tourette’s syndrome: effects of stimulant drugs. Neurology 1985; 35:1346-1348Crossref, Medline, Google Scholar
53. Price RA, Leckman JF, Pauls DL, Cohen DJ, Kidd KK: Gilles de la Tourette syndrome: tics and central nervous system stimulants in twins and nontwins. Neurology 1986; 36:232-237Crossref, Medline, Google Scholar
54. Singer HS, Rabins P, Tune LE, Coyle JT: Serum haloperidol levels in Gilles de la Tourette syndrome. Biol Psychiatry 1981; 16:79-84Medline, Google Scholar

