
A Randomized, Placebo-Controlled Dose-Comparison Trial of Haloperidol for Psychosis and Disruptive Behaviors in Alzheimer’s Disease
Abstract
Objective:The goal of this study was to compare the efficacy and side effects of two doses of haloperidol and placebo in the treatment of psychosis and disruptive behaviors in patients with Alzheimer’s disease.Method:In a 6-week random-assignment, double-blind, placebo-controlled trial (phase A), haloperidol, 2–3 mg/day (standard dose), and haloperidol, 0.50–0.75 mg/day (low dose), were compared in 71 outpatients with Alzheimer’s disease. For the subsequent 6-week double-blind crossover phase (phase B), patients taking standard- or low-dose haloperidol were switched to placebo, and patients taking placebo were randomly assigned to standard- or low-dose haloperidol.Results:For the 60 patients who completed phase A, standard-dose haloperidol was efficacious and superior to both low-dose haloperidol and placebo for scores on the Brief Psychiatric Rating Scale psychosis factor and on psychomotor agitation. Response rates according to three sets of criteria were greater with the standard dose (55%–60%) than the low dose (25%–35%) and placebo (25%–30%). The advantage of standard dose over low dose was replicated in phase B. In phase A, extrapyramidal signs tended to be greater with the standard dose than in the other two conditions, primarily because of a subgroup (20%) who developed moderate to severe signs. Low-dose haloperidol did not differ from placebo on any measure of efficacy or side effects. Conclusions:The results indicated a favorable therapeutic profile for haloperidol in doses of 2–3 mg/day, although a subgroup developed moderate to severe extrapyramidal signs. A starting dose of 1 mg/day with gradual, upward dose titration is recommended. The narrow therapeutic window observed with haloperidol may also apply to other neuroleptics used in Alzheimer’s disease patients with psychosis and disruptive behaviors. Am J Psychiatry 1998; 155: 1512-1520
Patients with Alzheimer’s disease often develop psychotic features (e.g., delusions and hallucinations) and disruptive behaviors (e.g., psychomotor agitation and physical aggression) (1–3). Psychosis and disruptive behaviors may be associated with a more rapidly dementing course (4–6). Psychomotor agitation may occur in nearly one-half of Alzheimer’s disease patients in outpatient clinics (7) and nursing homes (8), while aggressive behavior is less common (3, 9). The prevalence of delusions may range from 0% to 50% in Alzheimer’s disease (3, 10), but isolated symptoms (e.g., the belief that people are stealing things) are more common than typical psychotic disorders (1, 11). Hallucinations are rarely manifested early in the illness but may increase in prevalence as the illness progresses (3, 11).
Untreated psychosis and disruptive behaviors are distressing to patients and caregivers (12) and often lead to institutionalization (13, 14). Concomitant medical illness and the use of certain medications can induce or exacerbate these symptoms, and adequate medical management is important. The efficacy of nonpharmacologic interventions (e.g., behavior modification) remains to be established (15). Psychotropic medications, particularly neuroleptics, are widely prescribed for demented patients in outpatient, inpatient, and nursing home settings (16, 17). Earlier placebo-controlled studies of neuroleptics had flaws in research design that included diagnostic heterogeneity and the use of concomitant psychotropic medications (18–20). A meta-analysis of the extant studies suggested moderate superiority of neuroleptics over placebo in the treatment of psychosis and disruptive behaviors in dementia (21).
The elderly are prone to neuroleptic-induced neurologic side effects, particularly extrapyramidal signs and tardive dyskinesia (22). We previously showed that even moderate doses of neuroleptics (e.g., 5 mg/day of haloperidol) could not be tolerated by Alzheimer’s disease outpatients, primarily because of extrapyramidal signs (23). On the other hand, it is possible that the use of extremely low doses will lead to loss of efficacy. To guide clinical practice, dose-comparison studies are crucial to determine the dose range that leads to an optimal trade-off between efficacy and side effects.
Using a random-assignment, double-blind, placebo-controlled crossover trial, we compared the efficacy and side effects of “standard” (2–3 mg/day) and “low” (0.50–0.75 mg/day) doses of haloperidol in the treatment of psychosis and disruptive behaviors in outpatients with Alzheimer’s disease. The standard dose range was chosen to be toward the high end of doses used clinically but also to remain below the dose of 5 mg/day that often results in severe extrapyramidal signs (23). The low dose range was chosen to be at the lowest feasible dose that is administered in regular clinical practice. The main hypothesis was that the standard dose would be efficacious and superior to both the low dose and placebo, which would not differ from each other in efficacy. An additional hypothesis was that the standard dose would be associated with more extrapyramidal signs than the low dose or placebo, but that this level of extrapyramidal signs would not be intolerable and would not lead to study discontinuation at a greater rate among subjects receiving the standard dose than among subjects in the other two groups.
METHOD
All subjects were outpatients at a memory disorders clinic that is part of an Alzheimer’s disease research center. Subjects were required to meet the DSM-III-R criteria for dementia and the criteria for probable Alzheimer’s disease of the National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer’s Disease and Related Disorders Association (24). Diagnostic workup included neurologic and psychiatric evaluation, neuropsychological testing, laboratory tests, and a computerized tomographic (CT) or magnetic resonance imaging (MRI) scan of the brain. Exclusion criteria were drug or alcohol dependence, stroke (clinical evidence or a lesion of 2 cm or more in diameter on any CT or MRI slice), and a history or clinical evidence of other causes of dementia, including head trauma, Parkinson’s disease, Huntington’s disease, and multiple sclerosis. Patients who had had extrapyramidal signs before the onset of cognitive impairment were considered to have Parkinson’s disease and were excluded.
Patients who met the criteria for either psychosis or disruptive behaviors were eligible for the study. The criteria for psychosis required the presence of a delusion or hallucination as defined by the Schedule for Affective Disorders and Schizophrenia (SADS) (25), as well as a Brief Psychiatric Rating Scale (BPRS) (26) score of at least 4 (moderate severity) on the hallucinatory behavior or unusual thought content item, or a total score of 6 or more on these two items. The criterion for disruptive behaviors was a score of 4 or more (moderate severity) on the item for physical aggression or psychomotor agitation on the Behavioral Syndromes Scale for Dementia (7).
The research protocol was approved by the New York State Psychiatric Institute’s institutional review board. Informed consent was obtained from the patients and/or family members. The consent procedures followed specific New York State regulations concerning research involving patients who do or do not have the capacity to consent. Family members served as informants and were required to have had contact with the patient at least once a week on average during the 3 months before the patient entered the study.
Figure 1 summarizes the study design. In the initial 1-week single-blind phase, all patients received placebo. At the end of this week, the patients who still met the entry criteria were eligible for phase A, which was a 6-week random-assignment, double-blind, parallel-group, placebo-controlled comparison of three treatment conditions: standard-dose haloperidol (2–3 mg/day), low-dose haloperidol (0.50–0.75 mg/day), and placebo. To evaluate discontinuation effects, patients in the two haloperidol conditions in phase A received placebo in phase B, which also lasted 6 weeks. To evaluate replicability of the findings from the phase A haloperidol dose comparison, patients who received placebo in phase A were randomly assigned to one of the two haloperidol conditions in phase B (crossover). Raters and patients were blind to the study design throughout phase A and phase B. At the end of the trial, without breaking the blind, patients were treated openly with haloperidol or other medications (or received no medication) as deemed clinically appropriate.
The blinded research psychiatrist (D.P.D.) who evaluated the patients was also in charge of treatment, including dose adjustment, at all time points in the study. During both phase A and phase B, after the first week the daily dose was raised from two capsules (haloperidol, 2 mg or 0.50 mg, or placebo) to three capsules (haloperidol, 3 mg or 0.75 mg, or placebo). If side effects (e.g., extrapyramidal signs) were limiting, on the basis of the psychiatrist’s clinical judgment, the dose was maintained at two capsules daily. Therefore, in both phases, patients were on a stable dose for 5 weeks before the end-point evaluation of efficacy and side effects. Patients and family members were contacted frequently between visits to ensure compliance and to address any potential problems that could increase the risk of dropout, including lack of efficacy or the development of side effects.
All patients were free of psychotropic medications for at least 1 week before the initial 1-week placebo phase. Concomitant psychotropic medications were not prescribed during the study. No anticholinergic medications were used to treat extrapyramidal signs, as they can worsen cognitive deficits (27–29). Inability to tolerate side effects led to a lowering of the oral dose. In no case did the lowest permitted dose lead to intolerable side effects and study discontinuation.
The research psychiatrist (D.P.D.) conducted informant interviews with first-degree relatives, using the BPRS, the Behavioral Syndromes Scale for Dementia, and the items for psychosis and disorganization from the SADS, with the interval since the prior visit as the time frame of reference (the prior 6 weeks was used for the initial visit). The primary outcome measures of efficacy were the BPRS psychosis and hostile-suspiciousness factor scores, the Behavioral Syndromes Scale for Dementia item scores for psychomotor agitation and physical aggression (toward objects and/or others), and the sum of three target symptom scores identified from the psychosis and disorganization items of the SADS at study entry. The research psychiatrist also assessed systemic side effects with the Treatment Emergent Symptom Scale (30) (total score), extrapyramidal signs with the modified Targeting Abnormal Kinetic Effects (TAKE) scale (31) (total score), and tardive dyskinesia with the Rockland tardive dyskinesia scale (32). A neuropsychology technician administered the modified Mini-Mental State examination (33–35) to evaluate cognitive status. To assess functional impairment, the modified Blessed Functional Activities Scale (36) was completed by the informant.
The research psychiatrist administered all psychiatric rating instruments and completed side effect ratings. At the initial visit, severity of dementia was assessed with the Clinical Dementia Rating Scale (37); the BPRS, the Behavioral Syndromes Scale for Dementia, the psychosis/disorganization items of the SADS, and the Mini-Mental State were also administered. At the subsequent three major time points (1 week, 7 weeks, and 13 weeks), all instruments to assess efficacy and side effects were administered. Informant interviews were videotaped. A second psychiatrist (M.A.S.), who was trained in the use of these instruments but was not familiar with the study patients, rated a random subset of 20 videotaped interviews.
Blood for ascertaining haloperidol levels was drawn at the 7-week and 13-week time points, and clinical raters remained blind to the blood level results. A radioimmunoassay procedure (Janssen Pharmaceutica, Titusville, N.J.) was used to assay blood levels of haloperidol. The results of the radioimmunoassay for plasma haloperidol were strongly correlated with those from samples assayed by gas chromatography (r=0.96, slope=0.98, intercept=0.10 ng/ml). The sensitivity of the radioimmunoassay procedure enabled quantitation of 100 pg/ml with a root square deviation of less than 6%. Reduced haloperidol, which would have been at extremely low concentrations given the low oral doses used, was not assayed by this method.
For the statistical analysis, to achieve normal distributions, some variables were log transformed. Analyses were conducted on percent change scores. The primary analyses focused on the subjects who completed phase A, and significant multivariate analyses of variance (MANOVAs) were followed by univariate analyses. Intent-to-treat analyses were also conducted for the phase A sample, carrying forward the last observation. To confirm the phase A results obtained with percent change scores, analysis of covariance (ANCOVA) was conducted on the outcome measures at the end of phase A, with use of the corresponding measures at the start of phase A as covariates. Response rates in phase A were evaluated by chi-square analyses.
For phase B (crossover), between-group univariate analyses were conducted on the patients who received the standard dose or the low dose of haloperidol (after receiving placebo in phase A). In exploratory between-group univariate analyses, changes in efficacy measures in patients who received standard- or low-dose haloperidol in phase A (after 1 week of placebo) were combined with those of the corresponding standard- and low-dose haloperidol groups in phase B (after 7 weeks of placebo). For patients who received standard or low doses of haloperidol in phase A, within-group univariate analyses were conducted on the effects of phase B haloperidol discontinuation (switching to 6 weeks of placebo). All statistical tests were two-tailed.
RESULTS
Of the 71 Alzheimer’s disease outpatients in the study, 64.8% were female. The subjects" mean age was 72.1 years (SD=9.6), the mean duration of illness was 5.1 years (SD=2.9), and the mean number of years of education was 9.5 (SD=4.4). The ethnic breakdown was 56.3% white, 31.0% Latino, 11.3% African American, and 1.4% other. A history of past psychiatric disorder was uncommon (9.9%), and 35.7% of the subjects had a family history of dementia. Informants were mainly spouses (42.3%) or adult children (50.7%), and most (67.6%) lived with the patients. Forty-three patients (60.6%) had a Clinical Dementia Rating Scale score of 1 or 2 (mild or moderate dementia) and 28 (39.4%) had a score of 3 (severe dementia). The mean Mini-Mental State score (0=extreme deficit; 57=normal cognition) was 19.4 (SD=11.6), and the mean Blessed Functional Activities Scale score (0=no functional impairment;17=severe functional impairment) was 8.1 (SD=3.3). The mean BPRS total score was 59.2 (SD=9.4).
All 71 patients met the entry criteria for disruptive behaviors, and 51 patients (71.8%) met the entry criteria for psychosis. Physical aggression was moderate to severe in 49.3% (N=35), and psychomotor agitation was moderate to severe in 78.9% (N=56). One-way analysis of variance and chi-square analyses indicated that patients randomly assigned to the different cells did not differ in age, gender, total BPRS score, presence or absence of psychosis, and severity of dementia (Clinical Dementia Rating Scale and Mini-Mental State scores). Only 12 (16.9%) of the 71 patients had taken psychotropic medications during the month before study entry; three (4.2%) of these had taken low doses of neuroleptics on an as-needed basis.
Dropouts were defined as patients who did not complete a minimum of 3 weeks of treatment with complete end-point evaluation in phase A, the critical parallel-group comparison. Sixty of the 71 patients completed phase A. Of the 11 dropouts, five left the protocol during the pre-entry placebo period before random assignment to treatment. Of the six dropouts during phase A, four had been assigned to placebo, one to the low dose of haloperidol, and one to the standard dose. Eleven patients who completed phase A dropped out in phase B (crossover), leaving 49 who completed phase B. Dropouts in both phase A and phase B did not differ from the rest of the group in the main demographic or clinical features that were assessed.
The informant interviews at the 1-week, 7-week, and 13-week time points were videotaped. The second trained psychiatrist (M.A.S.), who was not familiar with the patients, rated 20 randomly selected videotapes without knowing the time points from which they came. There was good to excellent reliability between the original live ratings and the ratings by the second psychiatrist from the videotapes of the same interviews for the main efficacy measures (intraclass correlation coefficients=0.72 for BPRS total score, 0.54 for BPRS hostile-suspiciousness factor, 0.65 for BPRS psychosis factor, 0.80 for the sum of the target symptoms of the SADS item subset, 0.82 for physical aggression, and 0.92 for psychomotor agitation).
Sixty-six of the 71 patients completed the initial 1-week, single-blind placebo phase. For this phase, changes in efficacy measures were evaluated across the entire study group. BPRS total scores showed a mean 7% reduction (SD=13%) (t=4.17, df=65, p<0.001), the target symptoms of the SADS item subset showed a mean 11% reduction (SD=15%) (t=5.97, df=65, p<0.001), and psychomotor agitation showed a mean 1% reduction (SD=3%) (t=2.70, df=65, p<0.01). BPRS hostile-suspiciousness and psychosis factor scores and physical aggression scores did not change significantly. At the end of the initial 1-week placebo phase, all 66 patients still met the study entry criteria and were randomly assigned to treatments in phase A.
Phase A
Completer analyses
Sixty of the 66 patients completed phase A. At the end of phase A, 13 of 20 patients receiving the standard dose of haloperidol (2–3 mg/day) were taking three capsules daily (3 mg), 17 of 20 patients receiving the low dose (0.50–0.75 mg/day) were taking three capsules daily (0.75 mg), and all 20 patients receiving placebo were taking three capsules daily. Two patients in the standard-dose group had their doses reduced for short periods to 1 mg (one capsule) daily by their caregivers. However, for these two patients, the mean daily dose during the 6-week phase was less than 1.50 mg, and 2 mg was recorded as the phase A oral dose. Each treatment condition was analyzed as a single group, regardless of whether the daily dose was two or three capsules at the end of phase A.
Across the three treatment conditions, MANOVA on the five efficacy measures revealed a main effect of treatment group (F=3.23, df=2, 57, p<0.05). In the comparison between the standard dose of haloperidol and placebo, the MANOVA on the five efficacy measures revealed a main effect of treatment group (F=5.01, df=1, 38, p<0.04). In the comparison between the standard dose and the low dose, the MANOVA on the five efficacy measures also revealed a main effect of treatment group (F=5.05, df=1, 38, p<0.04). In the comparison between low dose and placebo, MANOVA did not reveal a significant effect of treatment group.
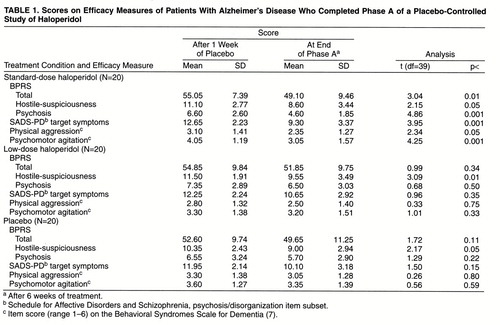
These analyses indicated that across the efficacy measures, therapeutic effects were greater with the standard dose of haloperidol than with either the low dose or placebo. The significant MANOVAs were followed by univariate analyses, and in the two-group comparisons, standard-dose haloperidol was significantly superior to placebo for BPRS psychosis factor scores (t=2.36, df=38, p<0.03) and psychomotor agitation (t=2.18, df=38, p<0.04) but not for the target symptoms of the SADS item subset (t=1.46, df=38, p=0.15), physical aggression (t=1.41, df=38, p=0.17), and BPRS hostile-suspiciousness factor scores (t=0.65, df=38, p=0.52).
The scores on extrapyramidal signs tended to be greater with standard-dose haloperidol than with placebo (t=1.82, df=38, p=0.08), but there were no significant differences in scores on the Treatment Emergent Symptom Scale, the Mini-Mental State, and the Blessed Functional Activities Scale. The trend for greater extrapyramidal signs with standard-dose haloperidol was caused by four patients whose scores increased by a mean of 18.25 points (range=13–24 points) on the 34-point modified TAKE scale, indicating moderate to severe extrapyramidal signs. For these four patients, baseline extrapyramidal sign scores were low (mean=2.75, range=0–4), and only two of these patients manifested hallucinations at baseline evaluation, making it unlikely that they had diffuse Lewy body disease and were misdiagnosed as having Alzheimer’s disease. Two of these four patients received haloperidol, 2 mg/day, and two received haloperidol, 3 mg/day. None of the other 16 patients taking the standard dose had an increase in extrapyramidal sign scores greater than 6 points; that is, they developed no extrapyramidal signs or only mild extrapyramidal signs. No patient in the low-dose and placebo groups had more than a 6-point increase in extrapyramidal sign score.
In phase A, standard-dose haloperidol was significantly superior to low-dose haloperidol for BPRS psychosis factor scores (t=2.12, df=38, p<0.05) and psychomotor agitation (t=2.51, df=38, p<0.03). There was no significant difference between the two groups in scores on the target symptoms of the SADS psychosis/disorganization item subset (t=1.80, df=38, p<0.08), the BPRS hostile-suspiciousness factor (t=0.16, df=38, p=0.87), and physical aggression (t=1.26, df=38, p=0.22). Extrapyramidal signs tended to be greater with the standard haloperidol dose than with the low dose (t=1.81, df=38, p<0.08), with no differences in scores on the Treatment Emergent Symptom Scale, the Mini-Mental State, and the Blessed Functional Activities Scale.
Low-dose haloperidol did not differ from placebo in any of the efficacy or side effect measures, even at trend levels.
To confirm the findings obtained with percent change scores, the same between-group comparisons were conducted with use of ANCOVA for the end-of-phase-A dependent measures with the corresponding start-of-phase-A measures as covariates. The results were similar to those obtained with percent change scores.
In phase A within-group analyses, patients taking the standard haloperidol dose improved significantly on all efficacy measures, while patients taking the low dose or placebo showed significant improvement only on the BPRS hostile-suspiciousness factor (table 1).
Intent-to-treat analyses
To address possible bias due to differential dropout, intent-to-treat analyses were also conducted for phase A, carrying forward the last observation. The dropout rate was relatively low, with four of six phase A dropouts being assigned to placebo and one each to the two haloperidol conditions. Since two-thirds of the phase A dropouts were taking placebo, the results from intent-to-treat analyses showed a slightly smaller improvement with placebo but virtually no change in the standard- or low-dose effects. Consequently, the results from the intent-to-treat analyses were virtually identical to those obtained in the completer analyses. In the intent-to-treat analyses, the standard dose of haloperidol was significantly superior to placebo with respect to percent change on the BPRS psychosis factor (t=2.53, df=49, p<0.02) and on psychomotor agitation (t=2.24, df=49, p<0.03) and significantly superior to the low dose on the BPRS psychosis factor (t=2.00, df=43, p=0.05) and on psychomotor agitation (t=2.47, df=43, p<0.02).
Response rates
Three sets of criteria for clinical response were evaluated. Response was defined a priori as a reduction of 25% or more in BPRS psychosis factor score, the target symptoms of the SADS subset items, or psychomotor agitation. Twelve (60%) of 20 patients taking the standard haloperidol dose met the response criteria on the BPRS psychosis factor, compared to six (30%) of 20 patients taking the low dose (χ2=3.64, df=1, p<0.06) and six (30%) of 20 patients taking placebo (χ2=3.64, df=1, p<0.06). Eleven (55%) of 20 patients taking the standard dose met the criteria for response on the psychosis/disorganization item subset of the SADS, compared to seven (35%) of 20 patients taking the low dose (χ2=1.60, df=1, p=0.20) and five (25%) of 20 patients taking placebo (χ2=3.75, df=1, p<0.06). Eleven (55%) of 20 patients taking the standard dose met the criteria for response on the symptom of psychomotor agitation, compared to five (25%) of 20 patients taking the low dose (χ2=3.75, df=1, p<0.06) and six (30%) of 20 patients taking placebo (χ2=2.56, df=1, p=0.11).
The changes in efficacy measures showed no significant correlations with the changes in extrapyramidal sign scores, both for the subjects taking standard-dose haloperidol and across the entire study group.
Phase B: Double-Blind Crossover
Eleven subjects who completed phase A did not complete phase B, leaving 49 patients for phase B completer analyses.
Patients in cells 3 and 4 received placebo in phase A and were randomly assigned to receive standard-dose or low-dose haloperidol in phase B. In these two cells, changes in phase B outcome measures were compared to evaluate the replicability of the advantage of standard dose over low dose that was observed in phase A (cells 1 and 2). In phase B, standard dose (N=8) showed nonsignificant superiority to low dose (N=10) for physical aggression (t=1.91, df=17, p=0.07), psychomotor agitation (t=1.70, df=17, p=0.10), the BPRS hostile-suspiciousness factor (t=1.60, df=17, p<0.13), the BPRS psychosis factor (t=1.32, df=17, p<0.21), and the target symptoms of the SADS item subset (t=1.66, df=17, p<0.12). In phase B, there were no differences between standard and low doses in scores on the Treatment Emergent Symptom Scale, the Mini-Mental State, and the Blessed Functional Activities Scale. Extrapyramidal sign scores were greater with the standard dose than with the low dose (t=5.00, df=17, p<0.01), and three of eight patients taking the standard dose had more than a 6-point increase in extrapyramidal sign scores. When results from phases A and B were combined, seven (25%) of 28 patients taking the standard dose had more than a 6-point increase in extrapyramidal sign scores.
Patients who received haloperidol in phase A crossed over to placebo in phase B (for completers: standard dose, N=17; low dose, N=14). To evaluate the effects of haloperidol discontinuation, these two groups could not be directly compared because the efficacy and extrapyramidal sign measures at the end of phase A differed between the two groups. Within-group analyses showed that the improvements with standard-dose haloperidol in phase A were partially reversed with placebo in phase B; there was a significant increase in psychomotor agitation (t=2.19, df=16, p<0.05), and there were nonsignificant increases in scores on the BPRS psychosis factor (t=2.07, df=16, p<0.06) and the BPRS hostile-suspiciousness factor (t=1.75, df=16, p<0.10) and in the sum of the target symptom scores of the SADS item subset (t=1.87, df=16, p=0.08). The increase in physical aggression was not significant (t=1.52, df=16, p<0.15). Extrapyramidal sign scores reverted to close to initial values (from mean=8.82, SD=8.06, to mean=3.59, SD=2.18; t=1.55, df=16, p<0.20), and there were no significant changes in scores on the Treatment Emergent Symptom Scale, the Mini-Mental State, and the Blessed Functional Activities Scale.
Patients in cells 1 and 2 received 1 week of placebo and then received either standard- or low-dose haloperidol in phase A. Patients in cells 3 and 4 received 7 weeks of placebo (1 week plus 6 weeks in phase A) and then received either standard- or low-dose haloperidol in phase B. These combined groups did not differ in efficacy measures at the end of the 1-week and 7-week placebo periods, respectively. Hence, exploratory analyses of efficacy measures in these combined groups were conducted, because the main phase A and phase B analyses were limited in statistical power. The combined group of 28 patients who received the phase A standard dose (cell 2) and the phase B standard dose (cell 4) were compared with the combined group of 30 patients who received the phase A low dose (cell 1) and the phase B low dose (cell 3). In these univariate analyses, the standard dose was significantly superior to the low dose for the BPRS psychosis factor (t=2.56, df=57, p<0.02), the target symptoms of the SADS psychosis/disorganization item subset (t=2.51, df=57, p<0.02), physical aggression (t=2.29, df=57, p<0.03), and psychomotor agitation (t=3.10, df=57, p<0.005) but not for the BPRS hostile-suspiciousness factor (t=1.25, df=57, p<0.25).
The research psychiatrist’s assessment with use of the Rockland tardive dyskinesia scale at the end of the 1-week placebo phase, the end of phase A, and the end of phase B indicated that no patient developed tardive dyskinesia during the course of the study, which involved a maximum 6-week exposure to haloperidol.
Haloperidol Blood Levels
Blood for determination of plasma haloperidol levels was drawn at the end of phases A and B. To enhance statistical power, in addition to phase A levels, phase B levels were ascertained for the two groups that crossed over from phase A placebo (haloperidol level=0 ng/ml) to phase B low- or standard-dose haloperidol.
Plasma haloperidol levels were detectable in all patients taking the standard dose (2–3 mg/day). For the low-dose patients whose haloperidol levels were below the limit of detection by the radioimmunoassay assay (N=7), the lower limit of assay detection was used as the value for the haloperidol level. Blood levels ranged from 0.1 to 0.8 ng/ml in the low-dose group and from 0.8 to 3.8 ng/ml in the standard-dose group. There was strong correlation between oral dose (0.50, 0.75, 2, or 3 mg/day) and blood level (r=0.70, df=51, p<0.001). There was the suggestion that compared with oral dose, plasma haloperidol levels showed somewhat stronger correlations with changes in measures of efficacy and extrapyramidal signs (table 2).
DISCUSSION
To our knowledge, this is the first published placebo-controlled dose-comparison study of neuroleptics used to treat psychosis and disruptive behaviors in dementia. In this group of Alzheimer’s disease outpatients, haloperidol in doses of 2–3 mg/day was efficacious and was superior to both low doses of haloperidol (0.50–0.75 mg/day) and placebo for specific outcome measures of both psychosis and disruptive behaviors. Similar results confirming the main study hypothesis were obtained in completer and intent-to-treat analyses, primarily because of the low dropout rate and the preponderance of placebo dropouts. For all efficacy measures, the differences between standard dose and low dose were as large as the differences between standard dose and placebo, confirming that the standard dose was indeed efficacious. The strength of these findings was reinforced by the efficacy advantage for standard-dose over low-dose haloperidol in phase B in the patients who crossed over from placebo in phase A. Low-dose haloperidol was indistinguishable from placebo for all efficacy and side effect measures, suggesting that doses below 1 mg/day are unlikely to be efficacious in the treatment of Alzheimer’s disease patients with psychosis and disruptive behaviors.
Response rates according to the three sets of responder criteria suggested superiority for the standard haloperidol dose (55%–60% response) compared with the low dose (25%–35%) and placebo (25%–30%). These differences are as large or larger than those reported in neuroleptic trials in schizophrenia (38–40). The findings are consistent with those from a study that compared flexible-dose thiothixene with placebo in a nursing home sample (41) but are greater than the effects reported in prior studies (21). A meta-analysis of these earlier studies of neuroleptics in agitated patients with any form of organic brain syndrome showed moderate superiority for neuroleptics over placebo (p=0.004, one-tailed). However, the effect size was reduced by the high placebo response rate in some studies (18, 20), raising the concern that the inclusion of patients with very mild symptoms (to enhance sample size) increased type II error. In contrast to our study, earlier studies permitted diagnostic heterogeneity in sample selection and the use of concomitant psychotropic medications during the trials (18–20).
In this study, both psychosis and disruptive behaviors improved with the standard dose of haloperidol. Scores on the BPRS hostile-suspiciousness factor improved with all three treatment conditions and showed the lowest interrater reliability among the five outcome measures. These factors may account for the failure to observe an advantage for the standard dose over the low dose or placebo on this measure. The majority of patients manifested both psychosis and disruptive behaviors, making it difficult to evaluate whether neuroleptics were more specific for the treatment of psychotic features than for disruptive behaviors (42). Larger samples would be needed to evaluate whether the presence of specific subtypes of symptoms predicts preferentially good response to neuroleptic treatment.
The subgroup of patients taking the standard dose who developed moderate to severe extrapyramidal signs (20% in phase A) did not meet published criteria for Lewy body disease (43, 44). However, autopsy of these patients was not obtained, and the possibility of Lewy body disease could not be excluded entirely. Currently, it remains unclear how patients susceptible to neurologic side effects can be identified in advance so that neuroleptics might be avoided. Neither haloperidol condition led to significant changes in general somatic side effects, cognition, or ability to perform activities of daily life. Overall, these data indicate that the use of haloperidol, 2–3 mg/day, leads to an acceptable trade-off between efficacy and side effects for Alzheimer’s disease patients with psychosis and disruptive behaviors, with the exception of patients who develop moderate to severe extrapyramidal signs. Long-term neuroleptic treatment is associated with the risk of developing tardive dyskinesia, particularly in elderly patients with schizophrenia (22). No patient developed tardive dyskinesia during the study, but neuroleptic exposure needs to be considerably longer than 6 weeks for systematic evaluation of this issue in patients with Alzheimer’s disease.
The newer atypical neuroleptics (e.g., risperidone, olanzapine, clozapine) have a low propensity for neurologic side effects and might be preferred for these patients (45). However, unlike haloperidol, these newer agents can cause orthostatic hypotension, with an increased risk of falls among the elderly (46). From a theoretical perspective, neuroleptics with prominent anticholinergic properties may worsen cognition, but this has not been empirically studied. Haloperidol has low anticholinergic properties (29), and Mini-Mental State scores did not differ between groups in this study. Additional neuropsychological tests were attempted but could not be completed by the majority of patients because of the severity of their dementia. Therefore, the possibility that the use of haloperidol may lead to subtle cognitive deterioration in Alzheimer’s disease cannot be excluded entirely (23). A recent study found that both the severity of persecutory ideas and the use of neuroleptics were associated with greater cognitive decline in Alzheimer’s disease (47). Currently, it remains unclear whether the presence of psychosis or the treatment for psychosis heralds a poor prognosis. Therapeutic trials with acetylcholinesterase inhibitors to treat the cognitive deficits of Alzheimer’s disease have been associated with an incidental finding of improvement in disruptive behaviors (48). Although this finding has not been firmly established, use of these agents may permit the concomitant use of lower doses of neuroleptics and other psychotropic medications to treat psychosis and disruptive behaviors.
In this study, patients who were switched after 6 weeks from standard-dose haloperidol to placebo showed signs of worsening symptoms, indicating that a longer period is necessary before discontinuation of medication is attempted. Recent longitudinal data from a large cohort of Alzheimer’s disease patients with mild to moderate disease (3) showed that psychotic features were moderately persistent over time. Disruptive behaviors, particularly psychomotor agitation, were highly persistent during several years of follow-up. These findings have implications for the optimal duration of treatment for specific behavioral complications before discontinuation is attempted. Continuation treatment studies with psychotropic medications in Alzheimer’s disease patients with psychosis and disruptive behaviors need to be conducted.
There has been little work examining the utility of monitoring neuroleptic blood levels in Alzheimer’s disease (49). In this study, haloperidol oral dose and blood levels showed a fairly strong correlation (r=0.70, p<0.001). The neuroleptic naivete of these Alzheimer’s disease patients (only three patients had received neuroleptics) and the short 6-week trial duration may have precluded the alterations in drug absorption and metabolism that occur with chronic neuroleptic treatment in schizophrenia, with the resultant distortion of the relations between oral dose and blood level (50, 51). The correlations between blood level and outcome measures of both efficacy and side effects appeared to be stronger than the associations between oral dose and outcome measures, but they were not strong enough to suggest clinical utility for blood level monitoring. Of note, the highest haloperidol level was 3.8 ng/ml, and clinical response occurred at levels that were always below the therapeutic window of approximately 5–15 ng/ml postulated for the treatment of schizophrenia in young adults (52, 53). In addition, moderate to severe extrapyramidal signs developed in some patients at these low blood levels. These data clearly show that the increased sensitivity to neuroleptics in elderly Alzheimer’s disease patients is not likely to be due to pharmacokinetic changes. A pharmacodynamic explanation—for example, degeneration of dopaminergic neurons leading to greater sensitivity to even low oral doses of neuroleptic medication—is more likely.
Overall, the results suggest a favorable therapeutic profile for haloperidol in doses of 2–3 mg/day and suggest that doses below 1 mg/day are ineffective. In the context of prior work indicating a high frequency of intolerable extrapyramidal signs at haloperidol doses of 5 mg/day or higher, these data indicate that there is a narrow therapeutic window for the use of haloperidol in Alzheimer’s disease patients. Given that a large subgroup receiving 2–3 mg/day of haloperidol developed moderate to severe extrapyramidal signs in this study, a starting dose of 1 mg/day with gradual upward dose titration is recommended, with close monitoring of the trade-off between efficacy and side effects. Although these results might be extrapolated to the use of other neuroleptic medications, empirical data are needed. With the newer atypical neuroleptics, the decreased propensity for neurologic side effects may make it possible to safely use even higher dose equivalents. However, several of these agents carry the risk of orthostatic hypotension, which can lead to falls and fractures. Clearly, comparison of the relative efficacy and propensity for side effects of different classes of neuroleptics and other agents requires head-to-head controlled trials in Alzheimer’s disease patients with psychosis and disruptive behaviors.
Received Jan. 29, 1998; revision received May 5, 1998; accepted May 12, 1998. From the Department of Biological Psychiatry, the Department of Analytical Psychopharmacology, and the Memory Disorders Clinic, New York State Psychiatric Institute; the Taub Center for Alzheimer’s Disease Research, New York; the Gertrude H. Sergievsky Center at Columbia University, New York; and the Department of Psychiatry, the Department of Neurology, and the Division of Epidemiology in the School of Public Health, College of Physicians and Surgeons of Columbia University, New York. Address reprint requests to Dr. Devanand, New York State Psychiatric Institute, 722 West 168th St., New York, NY 10032. Supported in part by grants MH-44176, MH-50038, and MH-55735 from NIMH; grants AG-07370, AG-07232, and AG-08702 from the National Institute on Aging; NIH grant RR-00645; and the Charles S. Robertson Memorial Gift for Alzheimer’s Disease Research from the Banbury Fund.
 |
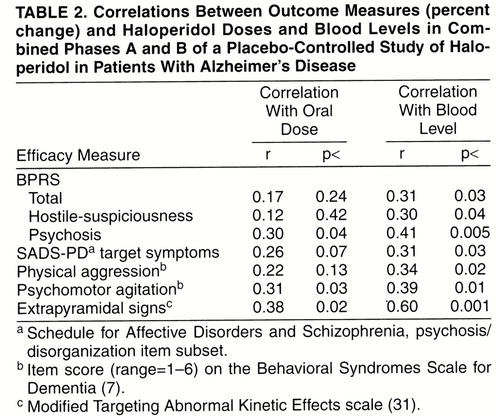 |

FIGURE 1. Design for a Placebo-Controlled Study of Haloperidol in Patients With Alzheimer"s Diseasea
1. Reisberg B, Borenstein J, Salob SP, Ferris SH, Franssen E, Georgotas A: Behavioral symptoms in Alzheimer’s disease: phenomenology and treatment. J Clin Psychiatry 1987; 48:9–15Medline, Google Scholar
2. Rubin EH, Morris JC, Berg L: The progression of personality changes in senile dementia of the Alzheimer’s type. J Am Geriatr Soc 1987; 35:721–725Crossref, Medline, Google Scholar
3. Devanand DP, Jacobs DM, Tang M, Castillo-Castaneda CD, Sano M, Marder K, Bell K, Bylsma FW, Brandt J, Albert M, Stern Y: The course of psychopathologic features in mild to moderate Alzheimer’s disease. Arch Gen Psychiatry 1997; 54:257–263Crossref, Medline, Google Scholar
4. Stern Y, Mayeux R, Sano M, Hauser WA, Bush T: Predictors of disease course in patients with probable Alzheimer’s disease. Neurology 1987; 37:1649–1653Crossref, Medline, Google Scholar
5. Drevets WC, Rubin EH: Psychotic symptoms and the longitudinal course of dementia of the Alzheimer type. Biol Psychiatry 1989; 25:39–48Crossref, Medline, Google Scholar
6. Rosen J, Zubenko GS: Emergence of psychosis and depression in the longitudinal evaluation of Alzheimer’s disease. Biol Psychiatry 1991; 29:224–232Crossref, Medline, Google Scholar
7. Devanand DP, Brockington CD, Moody BJ, Brown RP, Mayeux R, Endicott J, Sackeim HA: Behavioral syndromes in Alzheimer’s disease. Int Psychogeriatr 1992; 4:161–184Crossref, Medline, Google Scholar
8. Cohen-Mansfield J, Marx MS, Rosenthal AS: A description of agitation in a nursing home. Gerontology 1989; 3:M77–M84Google Scholar
9. Swearer JM, Drachman DA, O’Donnell BF, Mitchell AL: Troublesome and disruptive behaviors in dementia. J Am Geriatr Soc 1988; 36:784–790Crossref, Medline, Google Scholar
10. Reisberg B, Franssen E, Sclan SG, Kluger A, Ferris SH: Stage-specific incidence of potentially remediable behavioral symptoms in aging and Alzheimer’s disease: a study of 120 patients using the BEHAVE-AD. Bull Clin Neurosci 1989; 54:95–112Google Scholar
11. Wragg RE, Jeste DV: Overview of depression and psychosis in Alzheimer’s disease. Am J Psychiatry 1989; 146:577–587Link, Google Scholar
12. Small GW, Jarvik LF: The dementia syndrome. Lancet 1982; 11:1443–1445Crossref, Google Scholar
13. Rabins PV, Mace NL, Lucas MJ: The impact of dementia on the family. JAMA 1982; 248:333–335Crossref, Medline, Google Scholar
14. Haller E, Binder RL, McNeil DE: Violence in geriatric patients with dementia. Bull Am Acad Psychiatry Law 1989; 17:183–188Medline, Google Scholar
15. Leibovici A, Tariot PN: Agitation associated with dementia: a systematic approach to treatment. Psychopharmacol Bull 1988; 24:39–42Medline, Google Scholar
16. Beardsley RS, Larson DB, Burns BJ, Thompson JW, Kamerow DB: Prescribing of psychotropics in elderly nursing home patients. J Am Geriatr Soc 1989; 37:327–330Crossref, Medline, Google Scholar
17. Avorn J, Dreyer P, Connelly K, Soumerai SB: Use of psychotropic medication and the quality of care in rest homes. N Engl J Med 1989; 320:227–232Crossref, Medline, Google Scholar
18. Rada RT, Kellner R: Thiothixene in the treatment of geriatric patients with chronic organic brain syndrome. J Am Geriatr Soc 1976; 24:105–107Crossref, Medline, Google Scholar
19. Petrie WM, Ban TA, Berney S, Fujimori M, Guy W, Ragheb M, Wilson WH, Schaffer DJ: Loxapine in psychogeriatrics: a placebo and standard controlled clinical investigation. J Clin Psychopharmacol 1982; 2:122–126Crossref, Medline, Google Scholar
20. Barnes R, Veith R, Okimoto J, Raskind M, Gumbrecht G: Efficacy of antipsychotic medications in behaviorally disturbed dementia patients. Am J Psychiatry 1982; 139:1170–1174Link, Google Scholar
21. Schneider LS, Pollock VE, Lyness SA: A meta-analysis of controlled trials of neuroleptic treatment in dementia. J Am Geriatr Soc 1990; 38:553–563Crossref, Medline, Google Scholar
22. Jeste DV, Lacro JP, Gilbert PL, Kline J, Kline N: Treatment of late-life schizophrenia with neuroleptics. Schizophr Bull 1993; 19:817–830Crossref, Medline, Google Scholar
23. Devanand D, Sackeim H, Brown R, Mayeux R: A pilot study of haloperidol treatment of psychosis and behavioral disturbance in Alzheimer’s disease. Arch Neurol 1989; 46:854–857Crossref, Medline, Google Scholar
24. McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM: Clinical diagnosis of Alzheimer"s disease: report of the NINCDS-ADRDA Work Group under the auspices of the Department of Health and Human Services Task Force on Alzheimer"s Disease. Neurology 1984; 34:939–944Crossref, Medline, Google Scholar
25. Endicott J, Spitzer RL: A diagnostic interview: the Schedule for Affective Disorders and Schizophrenia. Arch Gen Psychiatry 1978; 35:837–844Crossref, Medline, Google Scholar
26. Overall JE, Gorham DR: The Brief Psychiatric Rating Scale. Psychol Rep 1962; 10:799–812Crossref, Google Scholar
27. Davies P, Maloney AJF: Selective loss of central cholinergic neurons in Alzheimer’s disease (letter). Lancet 1976; 1:1403Crossref, Google Scholar
28. Perry EK, Tomlinson BE, Blessed G, Bergmann K, Gibson PH, Perry RH: Correlation of cholinergic abnormalities with senile plaques and mental test scores in senile dementia. Br Med J 1978; 2:1457–1459Crossref, Medline, Google Scholar
29. Tune L, Carr S, Hoag E, Cooper T: Anticholinergic effects of drugs commonly prescribed for the elderly: potential means for assessing risk of delirium. Am J Psychiatry 1992; 149:1393–1394Link, Google Scholar
30. Guy W (ed): ECDEU Assessment Manual for Psychopharmacology: publication ADM 76-338. Rockville, Md, US Department of Health, Education, and Welfare, 1976Google Scholar
31. Wocjik JD, Gelenburg AJ, La Brie RA, Mieske M: Prevalence of tardive dyskinesia in an outpatient population. Compr Psychiatry 1980; 21:370–380Crossref, Medline, Google Scholar
32. Simpson GM, Lee JH, Zoubok B, Gardos G: A rating scale for tardive dyskinesia. Psychopharmacology (Berl) 1979; 64:171–179Crossref, Medline, Google Scholar
33. Folstein MF, Folstein SE, McHugh PR: “Mini-Mental State”: a practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 1975; 12:189–198Crossref, Medline, Google Scholar
34. Mayeux R, Stern Y, Rosen J, Leventhal J: Depression, intellectual impairment and Parkinson’s disease. Neurology 1981; 31:645–650Crossref, Medline, Google Scholar
35. Stern Y, Sano M, Paulson J, Mayeux R: Modified Mini-Mental State Examination: validity and reliability. Neurology 1987; 37(suppl 1):179–186Google Scholar
36. Blessed G, Tomlinson BE, Roth M: The association between quantitative measures of dementia and of senile change in the cerebral grey matter of elderly subjects. Br J Psychiatry 1968; 114:797–811Crossref, Medline, Google Scholar
37. Hughes CP, Berg L, Danziger WL, Coben LA, Martin RL: A new clinical scale for the staging of dementia. Br J Psychiatry 1982; 140:566–572Crossref, Medline, Google Scholar
38. Kane J, Honigfeld G, Singer J, Meltzer H: Clozapine for the treatment-resistant schizophrenic: a double-blind comparison with chlorpromazine. Arch Gen Psychiatry 1988; 45:789–796Crossref, Medline, Google Scholar
39. Marder SR, Meibach RC: Risperidone in the treatment of schizophrenia. Am J Psychiatry 1994; 151:825–835Link, Google Scholar
40. Gilbert PL, Harris MJ, McAdams LA, Jeste DV: Neuroleptic withdrawal in schizophrenic patients: a review of the literature. Arch Gen Psychiatry 1995; 52:173–188Crossref, Medline, Google Scholar
41. Finkel SI, Lyons JS, Anderson RL, Sherrell K, Davis J, Cohen-Mansfield J, Schwartz A, Gandy J, Schneider L: A randomized, placebo-controlled trial of thiothixene in agitated, demented nursing home patients. Int J Geriatr Psychiatry 1995; 10:129–136Crossref, Google Scholar
42. Raskind M, Risse S, Lampe T: Dementia and antipsychotic drugs. J Clin Psychiatry 1987; 48:16–18Medline, Google Scholar
43. McKeith IG, Perry RH, Fairbairn AF, Jabeen S, Perry EK: Operational criteria for senile dementia of Lewy body type (SDLT). Psychol Med 1992; 22:911–922Crossref, Medline, Google Scholar
44. McKeith IG, Galasko D, Kosaka M, Perry EK, Dickson DW, Hansen LA, Salmon DP, Lowe J, Mirra S, Byrne EJ, Lennox G, Quinn NP, Edwardson JA, Ince PG, Bergeron C, Burns A, Miller BL, Lovestone S, Collerton D, Jansen ENH, Ballard C, de Vos RAI, Wilcock GK, Jellinger KA, Perry RH, for the Consortium on Dementia with Lewy Bodies: Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the Consortium on DLB international workshop. Neurology 1996; 47:1113–1124Crossref, Medline, Google Scholar
45. Jeanblanc W, Davis YB: Risperidone for treating dementia-associated aggression (letter). Am J Psychiatry 1995; 152:1239Medline, Google Scholar
46. Chengappa RKN, Baker RW, Kreinbrook SB, Adair D: Clozapine use in female geriatric patients with psychosis. J Geriatr Psychiatry Neurol 1995; 8:12–15Medline, Google Scholar
47. McShane R, Keene J, Gedling K, Fairburn C, Jacoby R, Hope T: Do neuroleptic drugs hasten cognitive decline in dementia? prospective study with necropsy follow-up. Br Med J 1997; 314:266–270Crossref, Medline, Google Scholar
48. Raskind MA, Sadowsky CH, Sigmund WR, Beitler PJ, Auster SB: Effect of tacrine on language, praxis, and noncognitive behavioral problems in Alzheimer’s disease. Arch Neurol 1997; 54:836–840Crossref, Medline, Google Scholar
49. Dysken MW, Johnson SB, Holdon L, Vatassery G, Nygren J, Jelinski M, Kuskowski M, Schut L, McCarten JR, Knopman D, Maletta GJ, Skare S: Haloperidol concentrations in patients with Alzheimer’s dementia. Am J Geriatr Psychiatry 1994; 2:125–133Google Scholar
50. Sakalis G, Curry SH, Mould GP, Lader MH: Physiologic and clinical effects of chlorpromazine and their relationship to plasma level. Clin Pharmacol Ther 1972; 13:931–946Crossref, Medline, Google Scholar
51. Cheng YF, Paalzow LK, Bondesson U, Ekblom B, Eriksson K, Eriksson SO, Lindberg A, Lindstrom L: Pharmacokinetics of haloperidol in psychotic patients. Psychopharmacology (Berl) 1987; 91:410–414Crossref, Medline, Google Scholar
52. Volavka J, Cooper T, Czobor P, Bitter I, Meisner M, Laska E, Gastanaga P, Krakowski M, Chou JC, Crowner M, Douyon R: Haloperidol blood levels and clinical effects. Arch Gen Psychiatry 1992; 49:354–361Crossref, Medline, Google Scholar
53. Van Putten T, Marder SR, Mintz J, Poland R: Haloperidol plasma levels and clinical response: a therapeutic window relationship. Psychopharmacol Bull 1988; 24:172–175Medline, Google Scholar

